Team:BrockU/Description

Project Abstract
Flip recombinase (Flippase) is an enzyme that is capable of inducing site-specific recombination. When expressed, flippase has the ability to excise a sequence of DNA that lies between two specific recombinase cleavage sites, termed FRT sites. Flippase induces excision of the entire intervening sequence of DNA between these sites. The overall goal of our project is to engineer a light-activatable flippase by splitting the enzyme in two portions that each carry a genetically-encoded optogenetic tag. These optogenetic tags will allow for light-dependent induction of the association of the two flippase fragments, resulting in the reconstitution of flippase functionality. This construct, termed Opto-FLP, will allow us to control the function of flippase-mediated recombination precisely in space and time. Successful engineering of Opto-FLP will provide a light-gated system that can serve as a useful tool for a broad set of applications in cell biology and molecular genetics.
Testing the Functionality of Native Flippase
Before proceeding with the engineering of the desired optogenetic controlled flippase system, it was imperative to first test the functionality of native flippase to establish a control model. Flippase was inserted into the pCDF-Duet-1 vector. The vector was then transfected into BL21DE3 cells, which are a strain of competent E. coli cells that have a promoter for T7 polymerase, from which transcription can be activated in response to application of a common molecular biology reagent known as Isopropyl β-D-1-thiogalactopyranoside (IPTG).
In order to observe native flippase expression, we transfected our pCDF-Duet-1-FLP plasmid into BL21DE3 cells, along with the pKD4 plasmid. pKD4 is a plasmid that contains two FRT sites. By applying IPTG into a culture of these BL21DE3 dual-transfected cells, flippase will be activated and excise a portion of the pKD4 plasmid (Figure 1). Over time, this excised portion will be lost, as it does not contain an origin of replication, thus it cannot be passed on in a heritable manner to every daughter cell.
In order to observe native flippase expression, we transfected our pCDF-Duet-1-FLP plasmid into BL21DE3 cells, along with the pKD4 plasmid. pKD4 is a plasmid that contains two FRT sites. By applying IPTG into a culture of these BL21DE3 dual-transfected cells, flippase will be activated and excise a portion of the pKD4 plasmid (Figure 1). Over time, this excised portion will be lost, as it does not contain an origin of replication, thus it cannot be passed on in a heritable manner to every daughter cell.

Figure 1: Native Flippase Excision of pKD4. When transfected into BL21DE3 cells and activated using IPTG, flippase will excise a portion of the pKD4 plasmid. Two smaller plasmids will then be produced; one containing the ampicillin resistance gene and the origin of replication, and one containing only the kanamycin resistance gene. Due to the origin of replication, the plasmid carrying the ampicillin resistance gene will be passed on heritably to all daughter cells, while the plasmid with the kanamycin resistance gene will be lost over successive generations as it lacks an origin of replication.
We applied IPTG into a culture of dual-transfected BL21DE3 cells containing the pCDF-Duet-1-FLP and pKD4. The culture was left for 120 minutes after the addition of IPTG to observe the rate of flippase activity, with samples being taken every 30 minutes. In order to quantify flippase activity, the pCDF-Duet-1-FLP and pKD4 plasmids were isolated from the bacteria, and linearized by cutting the plasmids with the NotI restriction enzyme, then performing gel electrophoresis on these samples. At time point 0 min we expected that we would observe one band at 5.0 kB, corresponding to pCDF-Duet-1-FLP, and one band at 3.2 kB, corresponding to a complete pKD4. We also expected a faint band at band develop at 1.9 kB, corresponding to an excised fragment of pKD4 plasmid, which would be present due to leaky flippase expression. As time progressed, we would expect the band at 3.2 kB to become fainter, and the band at 1.9 kB to become brighter, as flippase should excise pKD4 into two distinct FRT-site containing plasmids. However, our results showed that the leaky expression of flippase resulted in recombination prior to introduction of IPTG (Figure 2). Although these results did not reflect the data we had anticipated, they demonstrated a necessity for optogenetic control of flippase, as native flippase cannot be fully temporally controlled at the transcriptional level using the IPTG-inducible T7 promoter system.

Figure 2: IPTG-induced Flippase Expression Results in the Excision of pKD4 at Varying Time Points. Dual-tranfected BL21DE3 cells containing pCDF-Duet-1-FLP and pKD4 were cultured overnight. IPTG was then added to the culture to induce flippase expression for a period of 120 minutes. It was expected that flippase would excise pKD4 more over time, however leaky flippase expression had excised all of the pKD4 plasmid prior to the induction of IPTG.
In silico Design of Opto-FLP
In order to avoid potential disruptions to flippase function, we have split flippase at a highly disordered region, which we hypothesize will increase the probability of optogenetic reconstitution of a functional flippase protein. These two portions termed FLPA and FLPB were conjugated to photosensitive proteins known as pMag and nMag, respectively. When the system is exposed to blue light (488 nm), pMag and nMag, which have been shown previously to specifically heterodimerize, will physically bring FLPA and FLPB together, resulting in reconstitution of an intact flippase protein, thereby stimulating flippase activity (Figure 3). Our hypothesis is that shining blue light on the system will render a functional flippase, allowing us to control its activity precisely in time and space. Each fragment (FLPA-pMag and nMag-FLPB) will be inserted into a separate multiple cloning site in the pCDF-Duet-1 vector. This will allow for ratiometrically equivalent expression of both halves of Opto-FLP.
We have chosen to use the pMag and nMag photosensitive tags as they heterodimerize and show complete specificity for each other. Other optogenetic systems have demonstrated issues with clustering or homodimerization. However, the magnet system contains a charged pair of optogenetic tags, one being positive, the other negative. Tagging each half of Opto-FLP with different optogenetic magnets ensures that one FLPA protein will associate with one FLPB protein.
We have chosen to use the pMag and nMag photosensitive tags as they heterodimerize and show complete specificity for each other. Other optogenetic systems have demonstrated issues with clustering or homodimerization. However, the magnet system contains a charged pair of optogenetic tags, one being positive, the other negative. Tagging each half of Opto-FLP with different optogenetic magnets ensures that one FLPA protein will associate with one FLPB protein.

Figure 3: Predicted Mechanism of Opto-FLP. By separating flippase into two halves and tagging each half with an optogentic magnet, we will be able to control flippase activity with blue light with spatial and temporal precision.
Molecular Cloning of pCDF-Duet-1-FLPA-pMag-nMag-FLPB
A typical cloned construct has one gene that is inserted into a digested vector. The Opto-FLP construct requires the insertion of multiple fragments into a vector, which can be completed using a process known as Gibson Assembly (Figure 4).
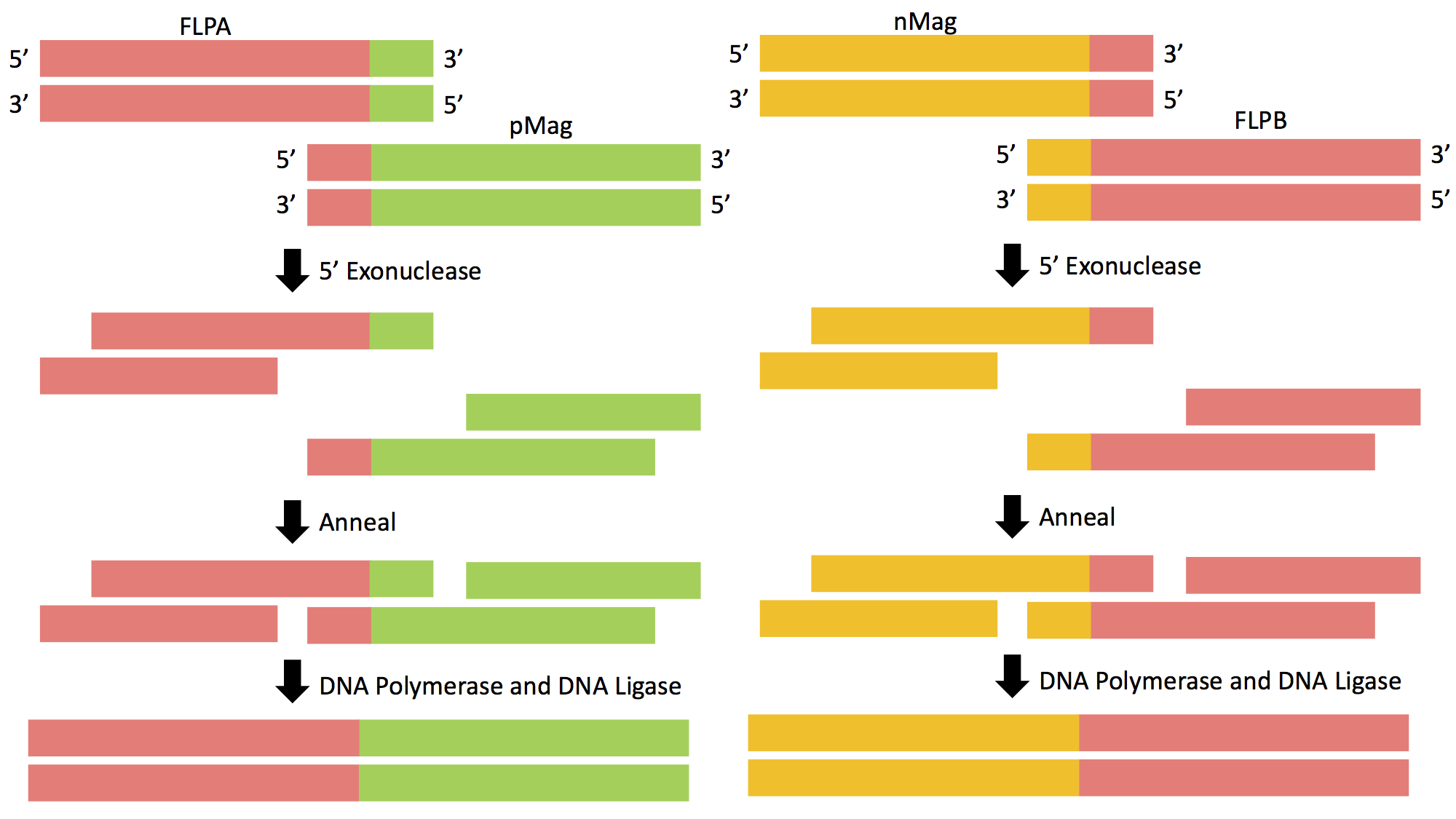
Figure 4: Gibson Assembly of FLPA-pMag and nMag-FLPB. The genetic sequence of Opto-FLP will be cloned into the pCDF-Duet-1 vector using Gibson Assembly in two separate reactions.
Prior to insertion, primers must be designed that roughly have 20 base pairs of homology to the gene of interest as well as homology to the vector itself. This will allow the complementary strands to be annealed, allowing the DNA fragments to be ligated together. There are three steps that allow for proper fragment insertion during Gibson Assembly. First, an exonuclease chews back DNA from the 5' end, revealing additional homology. Next, the resulting single-stranded regions on adjacent DNA fragments anneal to one another. This step is made possible by the exposed single stranded homologous ends created by the exonuclease. The final step entails a DNA polymerase incorporating nucleotides to fill in any gaps that were caused by the exonuclease or other forms of degradation. DNA ligase then covalently joins the DNA of adjacent segments, thereby removing any "nicks" in the DNA. The final product will be a circular vector now containing the genes of interest; in this case it will be the Opto-FLP construct in pCDF-Duet-1 (Figure 5).

Figure 5: Gibson Assembly Product of pCDF-Duet-1-Opto-FLP. By cutting pCDF-Duet-1 (A) in each multiple cloning site following each T7 polymerase promoter, the genetic sequence of each half of Opto-FLP could be inserted into pCDF-Duet-1 (B). This would allow for a ratiometric equal expression of both halves of Opto-FLP.
Proof of Concept of Opto-FLP Functionality
In order to demonstrate that our Opto-FLP construct is functional and is responsive to blue light, we will repeat our IPTG experiment. BL21DE3 cells transfected with pCDF-Duet-1-Opto-FLP and pKD4 will be cultured in a dark environment. At time 0, we will apply IPTG to th culture and expose the culture to blue light. We expect for there to be no flippase activity at time 0, since flippase would be nonfunctional in the dark. However, with the exposure of blue light, the two halves of Opto-FLP will associate, and excise the fragment from pKD4 (Figure 6).

Figure 6: Predicted Results of IPTG-Induced Opto-FLP Expression Resulting in the Excision of pKD4. Similar to the native flippase IPTG experiment, we would expect that there would be no flippase cleavage at time 0 min, as Opto-FLP would not be functional in the dark. Once subjected to blue light, we would expect Opto-FLP to gradually excise all the pKD4 in the bacterial culture.
Once we confirm that our Opto-FLP construct is functional, we will use a pGX-attp-FRT-STOP-FRT-GFP plasmid to demonstrate the functionality of our Otpo-FLP. Through dual-transfection of BL21DE3 cells with pCDF-Duet-1-Opto-FLP and the pGX-attp-FRT-STOP-FRT-GFP plasmid, we will be able to produce a system that can spatio-temporally control GFP expression. In a dark environment, Opto-FLP will not be active, thus there will be no GFP expression. If we subject the bacteria to blue light, we will be able to spatially control where we express Opto-FLP. Opto-FLP will excise the stop codons from the vector, allowing for GFP expression (Figure 7). This system will allow us to design patterns of GFP expression directly on bacterial plates to confirm the spatial precision of our Opto-FLP construct.

Figure 7: Opto-FLP Excision of STOP-GFP. Using our Opto-FLP construct, we will be able to control GFP expression in bacteria, exhibiting spatial control over flippase activity.