you better start swimmin'
Or you'll sink like a stone
For the times they are a-changin'
--Bob Dylan
The last years have seen a renaissance of research into Vibrio natriegens, the world’s fastest growing bacterium. V. natriegens has a lot of potential to become the next universally applicable chassis organism, challenging E. coli’s dominance. To fully realize this potential, several steps have to be taken. We intended to contribute to this coup d’état by providing the research community with open source strains, tailored specifically to cloning, protein expression and interaction. Additionally, we conducted basic microbial research, establishing procedures and protocols for cloning and molecular biology research.
Metabolic engineering relies heavily on iterative testing. To shorten the duration of these iterations means to enable broader and more in-depth acquisition of data.
Traditional cloning methods are limited by their rigidity and the time it takes to assemble constructs from multiple fragments. Recently, cloning methods based on type IIs restriction enzymes have addressed these issues. The synergy generated by combining these with V. natriegens would result in the world's fastest cloning platform, the Marburg Collection.
First isolated from salt marsh mud on Sapelo island off the coast of Georgia in 1958 (Payne et al. 1958), the gram-negative bacterium was first named Pseudomonas natriegens (Payne et al. 1960). These early studies also revealed a broad range of tolerated pH conditions with an optimum at a pH of 7.5 (Payne et al. 1960). One feature of this newly discovered organism became immediately apparent: The incredible doubling rate, first determined to be around 9.8 minutes (Eagon 1962). We found that doubling rates of around 7 minutes were possible (own Data).
Only much later, studies found several possible reasons for this. Firstly, as most Vibrio species, V. natriegens genome is split and distributed on two chromosomes. This means that replication can start at two origins of replication (Ori), resulting in more in parallel replication.

Additionally, it was shown that the gamma-proteobacterium has an increased rRNA activity. This is because of a greater number of rRNA operons which are additionally controlled by stronger promoters (Aiyar et al. 2002) when compared to E. coli . More rRNA means more ribosomes since rRNA synthesis has been shown to be the rate-limiting step in their assembly (Miura et al. 1981). Estimates for the number of ribosomes in V. natriegens in the exponential phase suggest around 115,000 per cell, while E. coli is estimated to have between 70,000 and 90,000 (Failmezger et al. 2018).
This gives V. natriegens higher protein expression and lets it create biomass more quickly. Could this also be exploited for the production of high-value proteins? Most likely.
It was soon discovered that V. natriegens would make a perfect example by which to effectively and harmlessly demonstrate the basic techniques of microbiology to students, taking full advantage of the immense doubling rate (Mullenger 1973; Delpech 2001). Experiments regarding population structure, UV stress, and simply bare growth can be observed in a much shorter time frame.
V. natriegens has a single stage lifecycle and does not form spores. The rod shaped bacterium does however possess a single polar flagella (Austin et al. 1978).

The rod-shaped bacterium occupies a special ecological nice in estuarine and salt marsh regions, where strongly fluctuating salt concentrations and nutrient availability create a challenging environment. In order to adapt to these challenges, V. natriegens can utilize a wide range of substrates to outgrow competitors, as well as remain in a low metabolic state for extended periods of time when the nutrient pool is depleted (Nazly 1980). Furthermore, it has the ability to quickly draw available phosphorous from its environs and store them in polyphosphate bodies (Nissen et al. 1987) for later utilization. Yet another way in which V. natriegens is adapted to its variable environment is the storage of carbon as polyhydroxyalkanoates (PHAs), biodegradable polyesters that are accumulated in intracellular granules that can be broken down under starvation conditions (Chien et al. 2007).
From the well oxygenated upper layer of mud down to the anoxic layers, V. natriegens is able to grow in many surroundings. These extremes are challenges to which V. natriegens found solutions. At the surface of the mud, as well as in open waters, the bacteria are exposed to strong UV radiation. As a response to this, the DNA damage repair systems are highly active (Simons et al. 2010), with significantly elevated expression levels of some parts of the system compared to E. coli , thus increasing DNA sequence integrity. By switching to fermentation, growth can be maintained under anaerobic conditions, allowing the colonization of the lower strata in the mud.
V. natriegens was shown to be a vital part of the marine ecosystem because it has the rare ability to fix atmospheric nitrogen under anaerobic conditions, thereby enriching its habitat and playing a crucial role as a provider of essential, bioavailable nitrogen (Coyer et al. 1996) under anaerobic conditions.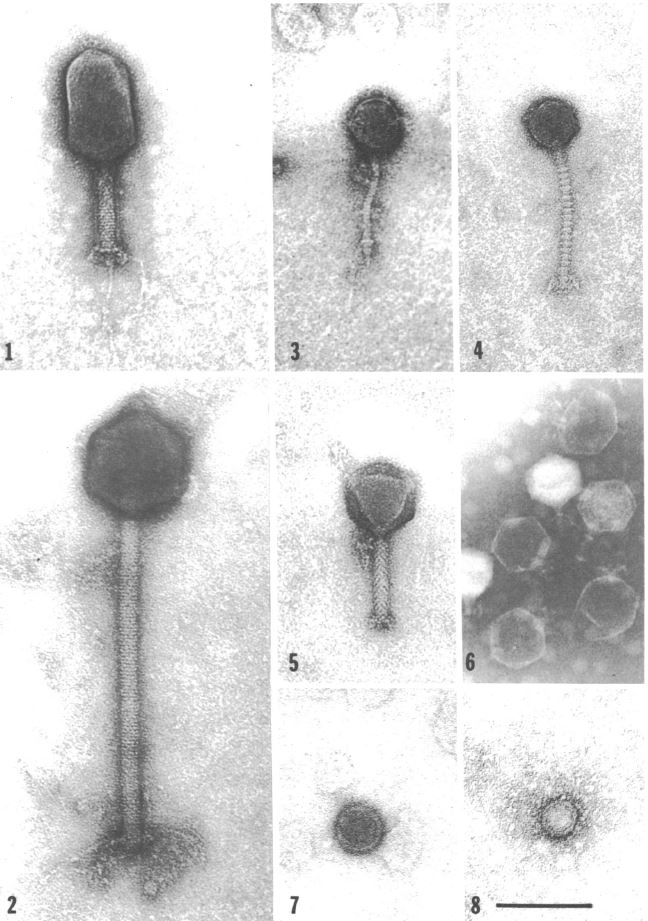
Unsurprisingly, V. natriegens has its own predators in its native habitat: Bacteriophages (Zachary 1974). Studies in the 70ies found a link between different phages and the prevailing environmental conditions (Zachary 1978) . This suggests the phages employ a divide and conquer strategy, specializing, thereby limiting competition while improving reproductive success. One phage may have adapted to be effective at lower temperatures, while others prefer higher temperatures. The same applies to other conditions like for instance salt concentration (Zachary 1976). Intriguingly, that opens the possibility of creating genetic parts derived from these phages, applicable to future work with V. natriegens, analogous to the T7 system in E. coli. On the other hand, one study could show, that the phage carrying the toxins associated with V. cholerae could not replicate in V. natriegens (Lee et al. 2016), further showing its harmlessness towards humans.
In contrast to that, some strains of Vibrio natriegens seem to be predators themselves. Confirmed as being pathogenic in several marine crustacea, most notably in the swimming crab Portunus trituberculatus, in which V. natriegens can lead to mortalities up to 85% (Bi et al. 2016). This is especially problematic since P. trituberculatus is farmed commercially in aquaculture in south-east China. The mechanism of that infection is poorly studied and it is not clear whether all strains of V. natriegens are pathogenic to crustaceans, but it is well known that many Vibrio species cause opportunistic infections in crustacea.
On a more positive note, a recent study could show that the sponge Aplysilla rosea develops a more diverse microbiome when exposed to V. natriegens (Mehbub et al. 2018). It can be speculated that this helps to protect the sponge from pathogenic microbes that then find it harder to colonize its surface.
A diverse, well-balanced metabolism allows a bacterium to grow in many conditions, while simultaneously making it interesting for biotechnological applications. V. natriegens can utilize a wide range of substrates (Hoffart et al. 2017), as well as grow in the absence of oxygen while being faster than other bacteria under the same conditions.
For many applications in which a modern laboratory work horse is expected to be useful, a high-resolution map of its metabolism is a prerequisite. One recent study generated just that (Long et al. 2017). By tracking 13C labeled carbon through the intricate network that constitutes V. natriegens metabolism, they were able to gain insight. Fore one, they found the it to be very similar to the well-studied E. coli metabolism. It has at the major catabolic and anabolic pathways, the same canonical amino acids and building blocks. Some notable differences between the two were the RNA content being 29% in V. natriegens compared to only 21% in E. coli, the presence of an enzyme for the decarboxylation of oxalate (Long et al 2017).
The specific uptake of many carbon sources per gram dry weight per time was shown to be significantly higher than in other comparable species of bacteria (Hoffart et al. 2017). Many commercially interesting compounds are already produced by unmodified V. natriegens. In the case of alanine production, only four deletions resulted in a strain that outperformed highly specialized E. coli and C. glutamicum production-strains by a factor of 9 to 13 times (Hoffart et al. 2017). Bioreactor based production on a large scale is expensive, therefore a strain generating similar or even higher yield per gram carbon source is highly desirable, improving temporal yield per fermentation unit.
When growing anaerobically, acids are produced that lead to an acidification of the medium but when grown under strong aeration, the pH can, depending on the medium, rise. This is most likely due to formation of ammonia (Eagon 1962).
The dependence on Na+ ions for its metabolic activity conveys a form of natural biocontainment, inhibiting growth in accidentally released V. natriegens (Webb and Payne 1971). The high salt content of the LBv2 media also presents possible contaminants with a barrier, reducing the likelihood of airborne microbes to take hold.

Recently, a study revealed that V. natriegens, surprisingly, is the most effective producer of selenium nanoparticles yet described (Fern`ndez-Llamosas et al. 2017). These particles with a diameter from 100-400nm have applications in diverse fields, such as medicine, microelectronics and more. Producing the nanoparticles in living systems has benefits over other methods, for instance, the low energy consumption as well as a coat of proteins preventing agglomeration of the particles. Also, V. natriegens exceptionally high resistance to selenite suggests possible applications in bioremediation of contaminated water and soils.
Working with V. natriegens
ATCC 14048 is the most commonly used strain of V. natriegens. Most results from earlier studies were generated from this strain. We recommend using ATCC 14048 in order to ensure that the findings will be applicable to your work and to adopt it as the standard strain. It is also the fastest strain known (Weinstock et al. 2016) so you won´t miss out on its incredible doubling time.
Stemming from subtropical regions, V. natriegens has not the same tolerance to low temperatures that we are used to from E. coli, but it is still much more resilient to cold than B. subtilis. At the root of that lies the low catalytic activity of V. natriegens native catalase at low temperatures, which is then unable to detoxify reactive oxygen species. To address that problem, scientists from SGI introduced homologous catalases, thereby creating a strain with comparable cold tolerance. With this adaptation, storage on plates can now be extended beyond the four weeks at room temperature by placing the plates in the fridge (Weinstock et al. 2016) In storage at -80°C in a standard 20% glycerol stock the cells stay viable almost indefinitely. And even when working with the wildtype, your cells won´t die overnight when placed at 4°C. Only after a week they will notably start decreasing in viability.
When working with V. natriegens ATCC 14048, no special precautions have to be taken since it has been shown that no known pathogenicity associated genes were present (Weinstock et al. 2016) Also, there is continued record of research since 1958 with not a single documented incident of a human infection with V. natriegens.
Lately, a new branch of microbiologists developed an interest in V. natriegens. Their main focus lies in establishing state of the art methods and bringing V. natriegens to its full potential. We see our project as part of this movement and wish to make V. natriegens easily accessible to researchers. One groundbreaking paper (Weinstock et al. 2016) established several methods to make V. natriegens genetically accessible, as well as characterizing some central genetic parts. This paved the way for more research into applying its potential.
If you want to introduce genetic material into V. natriegens, you have several tried and tested options to choose from. You could use electroporation, a quick and simple method and the one with the highest transformation efficiency. Electroporation can also be used on wildtype cells. Or you could use chemically competent cells, the transformation works very similar to E. coli heat shock transformation. But in order to achieve good efficiencies using chemically competent cells, a nuclease deletion strain is needed. And if you want to insert especially large constructs, you can also use conjugation with very good success ( (Lee et al. 2016; Weinstock et al. 2016).
A fully sequenced genome is also a prerequisite for many studies. Currently, several sequences are available (Lee et al. 2016; Maida et al. 2013), one of them generated by us. The genome has a size of approximately 5.17 Mb. Automated annotation was performed, revealing more of the genomic structure, as well as enabling the reconstruction of the codon usage profile (Lee et al. 2016).
The genome of V. natriegens is, like in many species of the genus, split into two chromosomes. Chromosome one contains 3.24 Mb, chromosome two 1.92 Mb.
Plasmids, Promoter, resistance
Usually, a big hurdle when changing your chassis organism is a very basic question: Will my constructs still work? Do I need different ori? Do I have to redesign everything from scratch? Fortunately, the most commonly used origins of replication, ColE1, p15A and pMB1 are maintained just fine in V. natriegens (Weinstock et al. 2016) We could observe some differences in copy number, which we quantified. You can find the corresponding data on our Part collection page. To retrieve these plasmids from the cells, we have tested a wide variety of commercially available kits and found that all of them worked, returning plasmid of the same purity and yield as for E. coli. Promoter commonly used in E. coli also lead to expression in V. natriegens. Expression levels are slightly different, but we also quantified these and you can find the results on our toolbox page. The same goes for other basic genomic parts like terminators and ribosomal binding sites.
Commonly, when very high gene expression is needed, for instance in protein overproduction, the phage derived T7 system is used. The T7 phage infects E. coli but it was also successfully inserted into V. natriegens and shown to drive protein expression (Weinstock et al. 2016) One way of generating even stronger, phage derived promoters could be using V. natriegens derived phage expression systems.
Every organism has its unique set of resistances to certain antimicrobial molecules. In order to successfully use a new organism, awareness of its individual tolerances is required to use antibiotics successfully for selection. We could show that chloramphenicol, carbenicillin, tetracycline and kanamycin worked, but at different concentrations from E. coli. We measured the V. natriegens specific concentrations and made them available on our strain engineering page.
Plasmids in absence of a selective pressure were only retained for a short time (Own Data). Curing of plasmids that have no longer a purpose is therefore quick and fairly effortless.
MuGENT - Recombineering
Investigations into alternative genetic tools were also undertaken, most notably MuGENT and Recombineering.
MuGENT takes advantage of V. natriegens natural competence by inducing the TfoX system. The TfoX system, which is known from many Vibrio species, enables uptake and genomic integration of DNA parts via homologous recombination (Pollack-Berti 2010). In most cases, growth on chitin was identified as the trigger which activated the system. Regrettably, the trigger is not known for V. natriegens. Using this system, a group of researchers characterized this system in V. natriegens, making it usable by placing it under the control of an inducible promoter. Additionally, they demonstrated its potential by increasing the natural poly-hydroxybutyrate (PHB) production 100-fold (Dalia et al. 2017).
Recombineering takes advantage of the fact that short DNA oligonucleotides, that can easily be delivered into cells by a plethora of ways, can be used to generate point mutations, knockouts and small modifications in the genome, as well as on plasmids. The homologous flanks required are relatively short, allowing for constructs under 100 bp. Having this system available in V. natriegens means that generating new strains and gene variants is easy and quickly implemented (Lee et al. 2017).
Cell Free Protein-expression Systems (CFPS)
Very recently, several groups have published their work about cell free protein expression systems (CFPS) in V. natriegens (Failmezger et al. 2018; Des Soye et al. 2018; Wiegand et al. 2018). Cell free systems offer the advantage of direct access to many cell components as well as fine control over the conditions in the reaction (Carlson et al. 2012). While these are true for all CFPSs, the advantage of creating such a system from V. natriegens is the much higher ribosome count (Failmezger et al. 2018).
Although the system works well when expressing plasmids from E. coli, it could be shown that expression was much higher when plasmids prepared from V. natriegens were used. Possible explanations are differences in methylation patterns, resulting in degradation of the plasmid by the system (Failmezger et al. 2018). All this could be done by using protocols very similar to well established methods for other bacteria.
Cre- lox
The Cre- lox system was also successfully implemented and shown to work consistently with 600bp homology flanks (Weinstock et al. 2016) Recycling of selection markers, like antibiotic resistances, as well as the removal of incorporated genes becomes easy and reliable.
Also interesting for future protein related applications is, that a putative secretion signal was identified (Weinstock et al. 2016)
Fun facts:
The epithet natriegens was chosen after the realization that salt is needed for it to grow, natrie stemming from the Latin word natrium meaning sodium and the Latin verb egere, meaning to need.
Natural Competence
 What happened with the genetic Information when cells lysis? The DNA will be released to the environment. But can it be reused there? Or is the hole information lost? Under certain circumstances other cells can take up the free DNA. This ability of cells is called competence. A special kind is the natural competence. These are bacteria that have genes in their genome encoded for proteins involved in the DNA recognition and uptake and in the integration in the genome.
What happened with the genetic Information when cells lysis? The DNA will be released to the environment. But can it be reused there? Or is the hole information lost? Under certain circumstances other cells can take up the free DNA. This ability of cells is called competence. A special kind is the natural competence. These are bacteria that have genes in their genome encoded for proteins involved in the DNA recognition and uptake and in the integration in the genome.
In 1928 natural competence was first observed by Frederick Griffith even if he did not understand the genetics behind (Griffith et al.1928) . He already recognized that information can be transferred from one bacterium to another and thus he set the starting point of a big era to understand the DNA exchange. Thanks many more great scientists researched in this area, we now know alot about natural competence and the ability of some bacteria to take up free DNA. However, as in almost every research, there are still many open questions.
One of the most famous Vibrio species is Vibrio cholerae. This bacterial species triggers the disease called cholera, which is well known. However, this bacterium is like many other vibrio species also interesting for many other genetic features. When you think about how he has achieved his pathogenic properties, it come into your mind that this bacterium is maybe capable to do extensive gene transfer. What we know is, that the most Vibrio species are natural competence, that means that these organism can take up linear DNA from the environment und this genetic material can used for nutrients, to repair the genome or to promote horizontal gene transfer when integrated it into their genome by homologous recombination
(Antonova et al.2015)
. There was identified multiple environmental cues sufficient to induce natural transformation. For the most Vibrio species the phenomenon of natural transformation is induced by Chitin
(Meibom et al.2005)
. In the natural aquatic environment of for example V. cholerae there are many chitinous shells of crustacean zooplankton which induces the expression of the competence regulator Tfox
(Meibom et al.2005).
For natural transformation several steps are necessary: At first the DNA has to be transported into the cell. After the DNA is bound to the cell, the Type IV Pillus (T4P) picks it up and transported it through secretion pores in the outer membrane into the periplasm. From there the DNA is translocated across the membrane by using a conserved membrane pore into the cytoplasm. If the DNA is arrived in the cell, it can be integrated by homologous recombination
(Mellet al.2014).
This process is dependent on some cellular structures, including type IV Pilli or related pseudopilli and many competence-related proteins and various regulator systems. The regulator Tfox activated the Type VI secretion systems (T6SSs) and that allows bacteria to inject directly into prey cell membranes or cytoplasm. It is a nanomachine that have many functions, one is a part of the natural competence regulon. Although much process has been made in this area, the molecular details of natural competence is not understood completely
(Wu et al.2015).
The Flp/frt system
There are several methods available to manipulate bacterial chromosomes. As the strain engineering subgroup, we had to decide which kind of strategy to use. Since iGEM means a lot of work in a very short time, we decided to look up for a method that further accelerates our project. We could use common plasmid based methods that rely on selection for plasmid excision followed by its loss. For Gram-negative bacteria these strategy often relies on counter selectable levan-sucrase enzyme, encoded by sacB (Donnenberg & Kaper 1991) . The excision can restore the genotype before insertion or result in an exchange between the chromosome and the plasmid-encoded copy of the modified gene. The disadvantage of this method is that it is time-consuming. First, you need to clone a plasmid and then horizontal gene transfer from a donor to V. natriegens would be necessary. Additionally, the excision of the plasmid can lead to the pre-insertion genotype.One of the big advantages of Vibrio species is their ability to take up free DNA from their environment by natural competence (Blokesch et al. 2005) , (Wollenberg et al. 2010) . Using this skill, plasmid independent genomic modification with linear DNA fragments can be performed in V. natriegens as well. To select for clones, which integrated those linear fragments at least one fragment containing an antibiotics resistance cassette is needed (Hayes et al. 2017) .
It would be a big disadvantage to create strains not compatible to some of your plasmids because of the antibiotics resistances incompatibility. The solution is, to design those antibiotics cassettes in a way they can be removed after integration. It was shown, that genomic modifications using recombinase-based methods like the Cre/Lox system are possible in V. natriegens (Hesek et al. 2016) . A similar method is the FLP/FRT system. FLP is a protein of the integrase family recombinases derived from the yeast Saccharomyces cerevisiae (Landy et al. 1986) , (Sadowski 1995)) . The recombinases of this family are sharing conserved amino acids that are responsible for the mechanisms of strand cleavage and ligation by the integrase members (Chen et al. 1990) , (Pan & Sadowski 1992) .
The FLP recognition target contains an eight bp asymmetric core that is surrounded by two 13-bp symmetric elements (5'GAAGTTCCTATTCtctagaaaGtATAGGAACTTC3'). The recombinase attaches to the top or bottom strand next to the core region, cleaves the DNA and promotes efficient recombination between two FRT recombination sites (Guarascio et al. 1982).

In general, Flp mediated recombination between these sites can lead to insertion, excision, inversion, and translocation of DNA, depending on the direction of the FRT sites (Figure 1). Using FRT sites that are oriented in the same direction, leads to Flp mediated deletion of the region in-between.
(Sadowski 1995).
This system works in Vibrio species like V. cholera or V. fischeri for rapid genetic modifications
(Blokesch 2012)
.
In order to establish a new and rapid method for genomic modifications in V. natriegens, we will use the FLP/FRT system to create our strains.
Abstract
The construction of stable bacterial strains for heterologous gene or pathway expression is usually done by chromosomal integration. Especially for the industrial application this has high priority, because continuous selection for plasmids is financial unsustainable. However, transcription levels vary between different chromosome regions. We therefore measured the growth of 15 different Vibrio natriegens mutants with integrated chloramphenicol resistance in different integration sites. Furthermore, we will characterize the gene expression in the integration sites by using the lac operon. By measuring its β-galactosidase activity the transcription level at this locus can be determined.
Introduction
Description Chromosomal Integration
Homologous recombination is a class of genetic recombination, in which a genetic exchange takes place between a pair of similar/ identical DNA sequences (Alberts et al., 2012) . Plasmids and linear DNA fragments, which carry sequences homologous to chromosome of the host strain, integrate into the host genome by homologous recombination following transformation (Cassey et al., 1991) (Cassey et al., 1991).
In order to achieve optimal flux throughput and omit unwanted intermediates, the expression of crucial genes in a metabolic pathway should lead to a protein stoichiometry that is as stable as possible (Dongmei Bai Flagfeldt et al., 2009) . This aspect is of special importance for industrial applications.
This can be achieved through chromosomal integration, as it rules out plasmid number fluctuations and the plasmid loss that sometimes occurs in bacterial strains.Another advantage of chromosomal integration is that it makes the use of antibiotics while cultivating obsolete, since there is no need to apply selective pressure to get mutants, saving money and optimizing the growth.
Position effects
Bryant and colleagues showed that the expression level of a gfp gene cassette varies ~300 fold, depending of its position on the E. coli chromosome. We assume that the potential integration sites, in Vibrio natriegens, also vary in their gene transcription. To rationally choose an optimal integration site rationally, potential integration sites must have their transcription levels characterized. The amount of transcription that will take place in an integration site is influenced by multiple factors.
- Common regulatory factors,
The local general enhancers and repressors, with for example recruit the RNA-polymerase to a nearby promotor or facilitate the formation of the open complex (Wu et al. 2017).
- DNA-methylation
It has been shown that the expression regulation of some genes is based on the DNA’s secondary structure caused by methylation. In E. coli GATC sites are methylated and can influence the transcription of nearby genes (Plasterk et al., 1983). We expect this to also be the case in Vibrio natriegens, so this must be considered when selecting an integration site.
- Superhelical density
Adjacent genes can cause transcription coupled DNA supercoiling, the strength of the supercoiling depends on the transcription rate and the length of the transcribed region (Liu and Wang, 1987) . It is also known that microorganisms possess negatively supercoiled domains. It has been shown that the strength of some promotors, can be affected by supercoiling (Sobetzko, 2016) . We expect integration sites that are close to highly transcribed regions to be affected by DNA-supercoiling and to exhibit unexpected transcription levels.
- Histone like proteins
One master regulator in bacterial cells are histone like proteins such as H-NS, they bind to the DNA and affect the nearby gene expression (Sobetzko, Travers and Muskhelishvili, 2012) (Sobetzko, Travers and Muskhelishvili, 2012). The effect of histone like proteins on our integration sites gene expression can vary extremely, depending on if and what kind of histone like proteins bind nearby.
- Distance to the origin of replication
Through multiforked replication some chromosomal region have multiple copies. It has been show that chromosomal integration sites close to the ori have a higher transcription rate in E. coli most likely due to the increased copy number of the gene (Sousa, De Lorenzo and Cebolla, 1997) . We assume that this will even more so be the case in Vibrio natriegens, as it replicates even faster and therefore has to do even more multiforked replication.
Experiment execution
Constructing of the parts for the chromosomal integration
For the chromosomal integration a construct was chosen, which includes the transcription unit for the chloramphenicol resistance and 1kb homology flanks on each side. The homology flanks were amplified by Q5 PCR from the place on the chromosome of V. natriegens we want to integrate at. The chloramphenicol resistance was amplified by Q5 PCR from the pYTK vector. After that the resistance fragment and a homology fragment for each side were fused by a Q5 fusion PCR. At the end we created 15 constructs for the integration at different places on the chromosome.
Integration of the parts into the chromosome
We introduced our fragments by natural transformation into Vibrio natriegens, because its high efficiency (Dalia et al., 2017). The integration in the genome appeared by homologous recombination. We verified a correct integration by colony PCR.
Growth measurement
The integration of the chloramphenicol may lead to growth deficits as it influences the transcription of neighbours gens. Therefor we conducted growth measurement were we always measured three cultures of the WT and three of the mutants with the inserted sequence. For every culture pre-culture was incubated at 37°C in a shaker until OD600= 0,4-0,6. From this culture we inoculated the main-culture to OD600~0,02 and began to measure the OD600 every 15 minutes.
There are different E. coli strains for a variety of applications. In every lab diverse strains are stored in freezers for methods like cloning, protein expression and protein interaction. And all of them are being used. For our project Vibrigens, we decided to establish the world’s fastest three lab strains suited for specific needs based on our V. natriegens wildtype strain. VibriClone, VibriExpress and VibriInteract. VibriClone is engineered to be a chassis for accelerated cloning. VibriExpress is engineered to be a chassis to speed up heterologous protein expression. VibriInteract is engineered to be a chassis for accelerated protein interaction studies based on the Bacterial Two-Hybrid system.



Why do we actually use E. coli for cloning? What are the reasons why E. coli is the one and only workhorse for molecular cloning? A big milestone in molecular biology was the first cloning in the 70's with the organism E. coli. Since this eventful step, the cloning has been constantly improved and accelerated. In addition, the strains of E. coli used for the cloning were adapted as best as possible to the requirements.
But what about the selected Chassi? Of course, E. coli is a good choice because it is genetically simple, has a fast growth rate and is now well studied. However, there are so many different microorganisms, maybe there is a species that is even better suited for cloning. We introduce to you the organism, it is Vibrio natriegens. Imagine you have all the advantages of E. coli for cloning, but much faster and easier. Through the transition, it would be possible for us scientists to complete cloning in one day instead of three and as you all can imagine, that would speed up the entire science. The only thing missing so far to use V. natriegens as cloning host is the optimization and the characterization. Exactly this point is our project. We want to make V. natriegens accessible and useful for you so that you have a great advantage for your important work by the changeover.
Exonucleases and Endonucleases

Vibrio cholerae, like V. natriegens, is able to pick up DNA from the environment. However, most of the DNA is degraded by the nucleases secreted by the microbial community. Some transformable material is mostly available and can be taken up by naturally competent cells. V. cholerae has two extracellular nucleases: xds and dns. It has been shown for V. cholerae that the transformation efficiency greatly increases when the two extracellular nucleases are knocked out. Thus, the DNA to be transformed into the organism is no longer degraded (Blokeschet al.2008) . In order to build a cloning strain of V. natriegens, it is also very important to knock out the nucleases. Otherwise, the DNAses would digest the DNA and it would not be possible to transform DNA.
RecA

KatG
The katG gene encodes an enzyme with a catalase and peroxidase activity. Peroxidases use hydrogen peroxide as a substrate. A special form is the Catalase, which is also KatG. Catalases are enzymes that catalyze the reaction of hydrogen peroxide to water and oxygen. The fact that they make hydrogen peroxide harmless has a protective effect on the cell. Many aerobic organisms including E. coli have a catalase because they contain flavoenzymes that contribute to peroxide formation (Fuchs 2017) . Due to the catalase activity it is possible to store E. coli at low temperatures, which is very useful in the laboratory. That's what we want for our VibriClone, too. Because we will have the working with VibriClone as comfortable and practical as possible.The lac operon


Nowadays the production of high protein amounts is important for biotechnological and industrial uses. One of the widely used expression hosts is E. coli in combination with the T7 expression system. For the last century, the performance of E. coli was hugely satisfying, except of issues like the aggregation of inclusion bodies and the insolubility of some proteins
(Villaverde & Carrio 2003).
However, in the last years, scientific progress is getting faster and faster and it is time to question if there is an alternative to E. coli, a host that is keeping up with the time?
Imagine you could start a day early in the morning performing a transformation with your construct. After several hours, you could inoculate colonies in liquid culture and induce the expression of your desired protein in an overnight culture. The second day you would be able to purify your protein.
Using V. natriegens as a new host with its impressive doubling time and the ability to grow to a much higher cell density will arise new possibilities for molecular and synthetic biology. In order to do so, we will design the VibriExpress strain.
“Creating a strain that is optimized for efficient production of high quality protein material is of high value for the drug discovery process.”this was the view of Dr. Serghei Glinca, the Chief Executive Officer of CrystalsFirst, a company that is specialized on protein-crystal based applications in drug discovery. Click here to learn more about our Integrated Human Practices project. The functionality of the T7 system in V. natriegens was already demonstrated and additionally it is pointed, that V. natriegens yields higher amounts of soluble protein than E. coli (Weinstock, Hesek et al.,2016).
But how is the commonly used T7 system working?
The T7 system
The system is based on the bacteriophage T7 RNA Polymerase, that shows a very high activity and synthesizes RNA at a rate several times that of E. coli. Besides, it is highly selective for its own promoter. Combined with the ability to inhibit selectively the host RNA polymerase with rifampicin (Hartmann et al., 1967) , it allows the exclusive expression of genes under the control of a T7 RNA polymerase promoter (Tabor S., 1985) . Due to these advantages, the gene of the T7 RNA Polymerase was integrated into the E. coli BL21 genome and is controlled by a lacUV5 promoter (Moffatt et al, 1986) , (Jun Lee et al., 2015) . The gene of the desired protein is introduced into the cell using a vector with a T7 promoter (e.g. pET vector, commercialized by Novagen). Incubating the cells with IPTG leads to the expression of the T7 RNA Polymerase. Finally, the Polymerase recognizes the T7 promoter and the expression of the desired protein is enforced (Figure 1) (Steitz et al., 1999) . To maximize the produced protein amount, proteases like Lon or OmpT can be knocked-out (Dunn et al., 1988) .

The Lon protease
The Lon protease derives its name from the phenotype of E. coli lon gene mutants that form long undivided filaments upon UV irradiation (Simson et al. 1964) . It is a component of the protein quality-control systems and degrades misfolded and unfolded proteins in an ATP dependent manner (Chung & Goldberg 1981) and is also involved in the degradation of certain regulatory proteins like SulA, which is a component of the SOS system and an inhibitor of cell division (Mizusawa & Gottesman 1983) , Huisman, (D'Ari et al. 1984)).The OmpT protease
The other deleted protease OmpT is an aspartyl protease localizing at the outer membrane of E. coli. It is suggested that OmpT is involved in the pathogenicity of E. coli. More than 30 years ago is was shown that OmpT catalyzes the activation of plasminogen to plasmin (Bowles et al., 1981) . This function seems to be physiologically relevant for the virulence of clinical E. coli isolates (Lundrigan & Webb 1992) and other pathogens like Yersinia pestis (Subrahmanyam et al., 1992) . Additionally the level of OmpT expression was shown to increase in response to the induction of recombinant protein overexpression (DeLisa et al., 2000) . For T7 expression strains, the deletion of this protease is urgently required because it was also shown that OmpT is degrading T7 RNA Polymerase (Grodberg & Dunn 1988) . According to NEB (New England Biolabs Ipswich, Massachusetts) strains that are deficient in both proteases Lon and OmpT are more amenable for the production of proteins
Our vision is to create a strain similar to E. coli BL21 and maximize the protein yield to an optimum by not only integrating the T7 system into V. natriegens but also deleting proteases to decrease protein degradation during the purification process. This way we will design an optimized strain for high-speed and high-level protein expression. Making VibriExpress accessible for everyone, we would like to accelerate your research and support you all, making the world a better place.
Proteins are part of the building blocks of life and fulfill a variety of essential functions. But what could one protein alone achieve? Mostly proteins interact with partners or in complexes to fulfill their function. A general goal of science is to understand all biochemical processes that enable life as we know it. Thus, the understanding of protein interaction partners becomes more and more important.
It is crucial to have methods investigating interactions between different proteins since most proteins do not act alone but in complexes with other proteins. This circumstance is of central interest since it can reveal a lot about the function of a protein and the processes that it is involved in. Using those methods, one can study interactions of specific proteins but it is also possible to screen for potential interaction partners using a library of genes. The latter one was already used by the iGEM team Bielefeld-CeBiTec in 2016. In their project they established their own method to do interaction assays to screen for proteins with an innate affinity for a specific target
(Click here to learn more about the Bacterial-Two-Hybrid assay developed by iGEM Bielefeld-CeBiTec).
This shows that interaction studies can not only be used in basic research but could also play an important role as an application in synthetic biology.
Protein interaction studies can be done by several methods:
- Biochemical Methods
- e.g. Co-immunoprecipitation
-
Biophysical and Theoretical Methods
- e.g. Fluorescence Resonance Energy Transfer (FRET)
-
Genetic Methods
- e.g. Two-Hybrid Systems
A lot of these methods still need a lot of time to be carried out. This can be done faster! Therefore we created our third strain: VibriInteract. A strain which can be used for protein interaction studies.
For Two-Hybrid Systems, the chassis organisms Escherichia coli and Saccharomyces cerevisiae are used in a Bacterial Two-Hybrid (B2H) and Yeast Two-Hybrid (Y2H) system, respectively. The Y2H assay was a novel genetic system at that time used to detect protein-protein interactions heterologously in yeast (Fields & Song, 1989). 9 years later, a new system was proposed. Instead of S. cerivisiae, this system uses E. coli as chassis organisms (Karimova et al., 1998). That this genetic test is carried out in E. coli greatly facilitates the screening as well as the characterization of the interacting proteins. Moreover, it accelerated this assay since E. coli has a much faster growth than S. cerivisiae. Although B2H assays are the easiest and fastest methods to test for interactions nowadays we think that they can be done faster. Using E. coli (which is already faster than using an Y2H system), the assay still needs 24 to 72 hours of incubation. Using V. natriegens, we can accelerate this assay to 8 to 24 hours of incubation. Paired with one day cloning we can speed up this assay from 7-9 days if your work in E. coli to 3 days if you work in V. natriegens. By this, we create our very own Two-Hybrid Assay: Vibrio Two-Hybrid (V2H).
| An ordinary workflow of a Bacterial Two Hybrid Assay | An ordinary workflow of a Vibrio Two Hybrid Assay |
|---|---|
| 3 days cloning (at best) 2 - 4 days first transformation 2 - 4 days spot assay |
1 day of cloning (at best) 1 day first transformation 1 day spot assay |
| 7-9 days in total | 3 days in total |
The Bacterial Two-Hybrid system is based on reconstitution of a signal transduction pathway relying on the adenylate cyclase (CyaA). CyaA is a key regulator in a lot of cells like e.g. E. coli. It catalyzes the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP) (Figure 2 A)
(Krupinski, 1991).
cAMP binds to the catalytic activator protein (CAP). This interaction effects a conformational change which leads to a higher binding affinity to DNA. The complex binds to the CAP site upstream of the promoter and can interact with the RNA polymerase
(Kolb et al., 1993).
This interaction increases the affinity of the polymerase to the promoter. The positive regulation boosts the expression of the gene by a factor of approximately 50 compared to gene expression without the presence of cAMP (Figure 1). To conclude, the cAMP/CAP complex is a positive regulator of e.g. the mal operon and the lac operon.

For B2H an E. coli strain lacking the cyA gene, which is encoding the catalytic domain of the adenylate cyclase and does not produce cAMP, is used. Consequently, cells can no longer utilize maltose or lactose. This phenotype can be rescued using the complementary T25 and T18 fragments of the catalytic domain of the adenylate cyclase toxin from Bordetella pertussis. Those fragments are cloned to genes of interest in plasmids called pKT25 and pUT18, respectively. Those fragments when splitted can no longer synthesize cAMP (Figure 2 B). If two hybrid proteins interact in this assay the two fragments of the catalytic domain come into close proximity and produce cAMP(Figure 2 C). The cAMP/CAP complex is formed again and regulates the mal or lac operon positively. Cells containing interacting constructs can utilize maltose and lactose again. Based on this, interaction of proteins can be screened using methods based on the lac operon or the maltose operon. For this, several different media are used:
- LBv2 agar plates supplemented with IPTG and Xgal used for Blue-White Screening if the lac operon is functional. You can read more about Blue-White Screening in the VibriClone part of our description.
- MacConkey agar plates supplemented with maltose to test if the mal operon is functional. When cells utilize maltose, acids are formed and cells turn red because of the pH indicator neutral red.
- M63 minimal medium agar plates supplemented with maltose to test if the mal operon is functional by screening for growth of colonies.

The first CRISPR-loci were discovered in 1987 in the E. coli strain K12 (Ishino et al. 1987) and the first description of a cas gene and their homologies to DNA-related enzymes appeared in 2002 (Jansen et al. 2002). Since then, more than 9000 research articles have been published about the different CRISPR/Cas subtypes and their application in various CRISPR based technologies (Adli 2018). The potential applications are almost limitless, ranging from new methods in research to industrial applications as well as human gene therapy. With CRISPR/Cas, it is possible to specifically switch off genes in a large number of prokaryotic and eukaryotic organisms, to integrate foreign DNA into the genome of an organism or to use the DNA-binding property for in vitro experiments (Hsu et al. 2014).

The picture shows the Cas9 assembled with the sgRNA and DNA. The scaffold of the sgRNA is illustrated in purple and the spacer in blue. The PAM is shown in red.

Golden Gate Assembly is a revolutionary cloning method published 10 years ago (Engler et al., 2008). since its publication it became very popular giving rise to many assembly strategies based on this system. All systems published over the years offered improvements over previous publications. Nevertheless, although there are many systems available – often specialized for certain organisms or classes – the development by far is not at an end. We worked on a novel toolbox and developed an improved, highly flexible system focused on prokaryotes including a high number of parts characterized for the novel upcoming chassis Vibrio natriegens.
The innovation given by Golden Gate assembly was the introduction of type IIS restriction enzymes used for DNA digestion. A current workflow at that time consisted of DNA restriction, gel purification and finally ligation of the single parts. For restriction different type II restriction enzymes were taken making it important to check for compatible buffers and getting a six base pair scar after ligation. By introducing type IIS restriction enzymes into this workflow the system was highly improved. This new method offered many advantages like I) an independent enzyme recognition site that is removed after restriction, II) restriction and ligation in one pot III) directional assembly by conscious design of the cleavage sequences IV) no buffer incompatibilities due to the usage of one enzyme cutting at several spots and still producing different cleavage sequences (Engler et al., 2008).

Based on this, many new systems arose offering the possibility of building complex constructs out of single genetic parts in a fast, simple and flexible way. Full libraries of standardized genetic parts were developed enabling the user to build full combinatorial libraries. The first system developed was the modular cloning system (MoClo) offering a new hierarchical cloning method of defined biological parts (Weber et al., 2011) and consequently setting a new standard over the years.
Here we summarize some Golden Gate based systems in a chronological manner looking at the improvements over the years and compare their advantages and disadvantages.
The modular cloning system was the first one proposing a standard for Golden Gate based assembly. This toolbox offers 5 types of modules designed mainly for eukaryotes. The modules are stored in level 0 acceptor plasmids derived from the pUC19 backbone with a spectinomycin resistance and a LacZα cassette as dropout for blue/white screening. Custom level 0 plasmids are assembled by flanking the sequences with BpiI recognition sites and setting a single restriction-ligation-reaction with the correspondent plasmid. Up to 5 level 0 modules are assembled in an acceptor plasmid with ampicillin resistance and a LacZα-dropout by restriction with BsaI to transcription units. 6 transcription units can be assembled using BpiI into level 2 multigene constructs containing a kanamycin resistance and a Cred-dropout. Alternatively Esp3I can be use to transfer the constructs to intermediary levels to reach higher levels for assembly of bigger constructs.
This toolbox is best for constructs up to level 2 plasmids. Higher levels can be reached through intermediary levels but need two restriction enzymes for the assembly. Furthermore it is important to know in which level the current constructs stand to avoid messing up the acceptor plasmids for subsequent levels.
The Golden Braid system was mainly developed with focus on plants. It picks up the idea of the MoClo system and improves it offering a looping system for reaching higher levels of assembly. Level 0 modules are stored in entry plasmids surrounded by BsaI restriction sites containing an ampicillin resistance and a LacZα dropout. This modules are assembled to transcription units in one of two level α entry plasmids with kanamycin resistance and a LacZα dropout. Here the transcription units are surrounded by BsmBI restriction sites. The transcription units can subsequently be transferred into level Ω entry plasmids with spectinomycin resistance and BsaI restriction sites surrounding the multigene constructs. Every level α plasmid is a precursor of a level Ω plasmid and vise versa. This way the system allows to loop over the plasmids building in theory infinite bigger constructs doubling their size on every round.
This system makes the assembly of higher levels much simpler using only 1 restriction enzyme. Nevertheless the plasmids’ size is just doubled from level to level.
The Golden Braid 2.0 system is an improvement of its prior system. Some minor changes were introduced to make the system simpler and more versatile. The basic modules were defined and classified in 11 classes building optional subclasses e.g. protein tags. The level 0 modules are stored in a universal vector (pUPD) surrounding the sequences with BsaI and BtgZI recognition sites cutting on the same position. The current acceptor plasmids α and Ω have been supplemented by reverse plasmids allowing the inversion of single transcription units. The fusion sites were simplified by merging unnecessary ones. ‘Superparts’ were introduced consisting of pre-assembled modules to reduce the number of fragments needed for the assembly.
This changes do not create a completely new toolbox but complements the system with some handy facilities improving it notably. However the system is still limited to a duplication of the plasmids size from level to level.
The yeas toolkit, also known as the Dueber toolbox, offers a Golden Gate based system adapted for yeast. The basic level 0 parts are classified in 8 types with optional subtypes. New basic parts are assembled into entry plasmids by restriction with BsmBI. For building level 1 cassettes at least 8 parts are assembled by a restriction-ligation step using BsaI. The innovation of this toolkit compared to the previously reviewed is the use of connector sequences for level 1 and higher assembly steps. This way plasmids for yeast can be built de novo without the need of a defined backbone. Furthermore they integrated a method for simple chromosomal integration by lenearization of the plasmids with NotI. On top of this the connectors can be used as homology sequences e.g. for ligation-independent cloning, Gibson assembly, ligase cycling reaction or yeast in vivo assembly. Although this toolkit focuses mainly on yeast it can be used for working with E. coli as well. Unfortunately this toolkit does not arrange to any standard becoming incompatible with other toolkits.
The PhytoBrick system does not offer another genetic toolbox but a wide standard compatible with popular systems like GB 2.0 and MoClo aiming to create a standard focused primary on plant engineering efforts. The iGEM competition already accepted it as an standard and offers support for building parts designed for plants, yeast and bacteria. This system proposes 12 defined fusion sites applicable for the different genetic modules. The fusion sites are divided into three major classes for promoter parts, transcribed regions and terminator parts. This classes are divided into subclasses giving the flexibility to use optional modules like tags, promoter regulators and enhancer regions. The system also proposes two types of universal acceptor plasmids (UAPs) derived from the pSB1C3 plasmid where level 0 modules can be inserted by a single restriction-ligation step with BpiI or BsmBI respectively.
The Cross-disciplinary Integration of Design Automation Research lab (CIDAR) MoClo Library represents the first toolbox adapted for bacteria focused on E. coli. It offers 95 genetic modules for bacteria and proposes a modified protocol reducing the reaction time of the assembly steps from 5 hours to just 90 minutes. The toolkit provides a series of level 0 plasmids derived from the iGEM pSB1A2 backbone. Four modules are assembled into a series of level 1 destination plasmids derived from pSB1K3 to transcription units using BsaI. In a next step this transcription units can be assembled to multigene modules by restriction with BbsI. Up to 4 transcription units can be assembled to a level 2 multigene construct.
EcoFlex is a toolbox specialized on the use with E. coli that offers a wide variety of modules. It features a library of constitutive promoters, T7 expressions, RBS variations, synthetic terminators, protein purification tags and fluorescence proteins characterized in vivo and in vitro. Furthermore it is compatible with MoClo, Golden Braid 2.0 and PhytoBrick standard.
One unique feature of this kit is the ‘entry vector’ for ORFs. The parts are flanked by NdeI and BamHI sites again flanked by BsaI restriction sites. The NdeI sites provide the fusion sites for Golden Gate assembly without addition of amino acids to the coded protein and in combination with BamHI additionally providing compatible transfer to the commercial available pET vectors. If NdeI or BamHI are present in the coded protein NcoI and BglII can be used respectively for the assembly.
The second unique feature of EcoFlex is the inclusion of a secondary module. Here level 2 transcription units (TUs) can be subcloned into a corresponding secondary module using compatible BpiI sites located upstream the primary MoClo BsmBI sites. This way a module of multiple TUs can be ligated into a secondary module containing further TUs. The introduction of this feature offers a simplified and faster method for combinatorial pathway optimization.
Loop assembly provides a new toolbox designed for plant engineering purposes. It contains a series of 8 plasmids derived from pGreenII backbone divided in two sets to loop over several levels of assembly. Level 0 modules are assembled into odd receiver plasmids (pOdd) with a kanamycin resistance by restriction with BsaI. Four odd level transcription units are assembled into one even level receiver plasmid (pEven) with a spectinomycin resistance by restriction with SapI. The system can be elevated to higher levels by looping over odd and even receiver plasmids exponentially by the factor of 4. For the case that less than 4 modules are needed spacer plasmids are offered of 200bp random DNA free of BsaI or SapI recognition sites to close the gaps. The level 0 parts’ fusion sites are designed using the PhytoBrick standard previously reviewed and are therefore compatible with GB 2.0 and MoClo. A further improvement of this system is the use of unique nucleotide sequences (UNS) set between single transcription units. This UNS allow highly efficient PCR amplification of single functional units and allow the assembly into the plasmids by overlap assembly methods like Gibson assembly as well.
Although this is an improved and very simple but useful system the usage of SapI delivers a 3 bp fusion site restricting the number of possible fusion sites to a lower number and simultaneously downgrading compatibility with previous systems.
| Toolkit | MoClo | Golden Braid | Golden Braid 2.0 | MoClo – YTK | PhytoBrick | CIDAR MoClo | EcoFlex | Loop Assembly |
|---|---|---|---|---|---|---|---|---|
| Year of publication | 2011 | 2011 | 2013 | 2015 | 2015 | 2015 | 2016 | 2018 |
| Restriction sites | BsaI, BpiI (Esp3 for intermediary levels) | BsaI, BsmBI | BsaI, BsmBI (BtgZI as BsaI alternative) | BsaI, BsmBI (NotI for chromosomal integration) | BsaI, BsmBI (BpiI for compatibility with MoClo) | BsaI, BbsI | BsaI, BsmBI (NdeI + BamHI for secondary modules) | BsaI, SapI |
| Dropouts for negative selection | LacZα, CRed | LacZα | LacZα | sfGFP (only lvl 0) | Depends on the system respectively | LacZα | LacZα, RFP | LacZα |
| Resistances | Spectinomycin → ampicillin → kanamycin | Ampicillin → kanamycin → spectinomycin | Ampicillin → kanamycin → spectinomycin | chloramphenicol → ampicillin → kanamycin | Chloramphenicol, ampicillin, kanamycin, spectinomycin | Ampicillin → kanamycin | Chloramphenicol → β-lactamase | Kanamycin → spectinomycin |
| Target organisms | Mainly eukaryotes | Plants | Plants | Yeast | Plants | Bacteria | E. coli | Plants |
| Special features | First Golden Gate Assembly standard | Theoretically infinite looping over two sets of plasmids and levels | Flexible direction inversion of single TUs during assembly steps, introduction of classes and subclasses | Connector-based de novo plasmid assembly for yeast, chromosomal integration, | Not a toolbox but a proposal for a standard widely compatible with popular systems, accepted standard for the iGEM competition | First system focused on bacteria, enhanced protocol for Golden Gate reaction in 90 minutes | Widely versatile toolbox, adapted for E. coli, designed for circuit design, combinatorial pathway optimization, recombinant protein purification | Simple usage, wide compatibility due to PhytoBrick standard, usage of UNS |
| Enhancement per level | 6 TUs, but capped at level 3 | 2n | 2n | Depends on the system respectively | 4n | Up to 5 TUs on level 2 and up to 20 on level 3 | 4n | |
| Number of parts | 95 | 94 | 96 | 93 | 78 | 58 |
Our Goal
Our goal is to accelerate metabolic engineering by establishing a workflow for fast pathway construction, product screening and pathway optimization. Metabolic engineering is a broad field in synthetic biology, where existing pathways are divided into modular parts and then used and combined to build new pathways (Stephanopoulos et al.2012) . One of the best examples for the impact of metabolic engineering on solving global problems is the engineered production pathway for artemisinic acid, the precursor for a drug against malaria (Ro et al.2006) . In the old days the drug had to be isolated out of the plant Artemisia annua. This was very inefficient because of the limitations of plant growth and low yields of artemisinic acid. Therefore, the group of Jay Keasling engineered a metabolic pathway to produce the drug in yeast cells, which grow faster and produce a lot more artemisinic acid. As in this example, metabolic engineering enables the production of drugs and other valuable products with an increased yield when compared to product isolation from natural sources. Furthermore, the large variety of usable enzymes, pathways and chassis enables more flexibility in pathway design compared to chemical approaches.
Why should we accelerate metabolic engineering?
Although much progress in metabolic engineering was made in the last 20 years, there are still many drawbacks and limitations. For instance, it is crucial to have a big set of genes as combinable parts to find the optimal pathway and they have to be predictable. With many of them just poorly characterized, rational pathway design remains difficult. Our understanding of the working and interactions of enzymes at the level of the metabolism is still rudimentary. Consequently, metabolic engineering consists of much trial-and-error and researchers have to endure series of failures. One strategy to face these issues is modeling of enzyme behavior but currently limitations in computing power prevent this from being used in high-throughput. Another strategy is to use chassis as workhorses to test a lot of enzyme combinations and pathway variants. Here, organisms like E. coli, S. cerevisiae or C. glutamicum are often used but product formation is limited by growth rate and nutrient uptake (Stephanopoulos et al.2007). When a pathway is constructed and to be tested, one has to wait until the production strain has grown and then product formation is still limited by substrate uptake. Another big drawback is that screening for product often is expensive, time consuming and complicated. Conventional methods like LC-MS need a lot of time and resources and it is hardly possible to use it in high-throughput, for instance for a whole pathway library. To overcome these limitations and accelerate metabolic engineering, we established V. natriegens as chassis for metabolic engineering. Its doubling time of under 10 minutes makes it the perfect organism to quickly test many pathway variants or enzyme versions. Additionally, it has a glucose uptake rate of 3.90 g g-1h-1 while E. coli, S. cerevisiae and C. glutamicum have much lower rates (1.90, 3.52 and 0.37 g g-1 h-1, respectively). Coupled with the short doubling time, V. natriegens is a powerful chassis not only for research purposes but also for industrial usage, where high productivity is crucial.
How are we accelerating metabolic engineering?

3-Hydroxypropionic acid 3HPA
As a proof of concept for our engineering workflow we chose the biological production of 3-Hydroxypropionic acid ( 3HPA ). 3HPA is a compound of high industrial value.
In 2004 the U.S. Department of Energy recommended 3HPA as alternative to fossil oils for the chemical industry
(Werpy et al.2004).
3HPA as a platform chemical can be converted into many other compounds like acrylic acid and acrylamide, which both are also precursors for further compounds.
According to an estimate of 2014, acrylates alone have an annual market value of USD12 billion, making its precursor 3HPA an optimal target product for our metabolic engineering approach.
By finding the optimal pathway to produce 3HPA we could show the efficiency of metabolic engineering in V. natriegens as well as getting a big step closer to an alternative to fossil oils by biological, renewable resources.

Our pathway
In recent years many people concentrated on producing 3HPA via many different metabolic pathways
(Valdehuesa et al.2013)
;
(Vidra et al.2017).
Most of these pathways use glycerol or glucose as starting substrate, but there are also publications where acrylic acid, CO2 or uracil were converted to 3HPA
(Vidra et al.2017).
One promising route starts with glycerol which is dehydrated and oxidized.
For E. coli, a productivity of 6.6 mmol g-1 cdw h-1 has been shown
(Raj et al.2008).
However, this route depends on vitamin B12, which often can’t be taken up or produced by the chassis.
Many other routes start with glucose, which gets metabolized via propionate, lactate, β-alanine or malonyl-CoA
(Vidra et al.2017).
We decided to use a pathway, based on the conversion of acetyl-CoA into malonyl-CoA and finally 3HPA . According to
(Valdehuesa et al.2013)
who evaluated many pyruvate-derived production pathways from a thermodynamic point of view, this is one of the most efficient routes for 3HPA production.
This pathway was first established in E. coli, but it was also tried in many other chassis like S. cerevisiae and S. elongatus
(Kildegaard et al.2016);
(Liu et al.2017);
(Rathnasingh et al.2012).
We chose this route for several reasons.
Firstly, it is based on glucose degradation and V. natriegens has an unbeaten glucose-uptake rate, enabling high glucose consumption. Secondly, vitamin B12 is not necessary, the only cofactors involved are NADPH and biotin, both of which occur naturally in the organism.
Thirdly, theoretically just one further enzyme is needed to complete the pathway. All enzymes necessary for the production of malonyl-CoA are already present in V. natriegens and we just have to integrate the last enzyme, malonyl-CoA reductase.
Nevertheless, overexpression of acetyl-CoA carboxylase increases production, so we took both enzymes into consideration.
(Liu et al.2017)
proposed, that there are many ways to further optimize the pathway and direct the flow towards a high product titer.
Hence, implementing many variations of this pathway into V. natriegens and testing them in a fast manner promises a wealth of knowledge about pathway optimization and general principles of metabolic engineering.


One major goal of synthetic biology is to make biology engineerable and predictable. Currently metabolic engineering does still involve a lot of trial and error. With the number of possibilities for pathway finetuning this scales very fast, becoming very difficult to tackle with traditional methods.
Also, once we create such a library of metabolic pathway variants, possibly including thousands of strains, we have to find the most promising ones among these. A traditional approach would be to check the production of each strain in its own culture flask. Using for example HPLC or mass spectrometry.
A more suitable, easy way to quickly screen for metabolite concentration in a semi quantitative way are biosensors. One of the easiest to use classes of biosensors are metabolite-responsive transcription factors
(Liu et al. 2018).
They are usually very specific towards their distinct substrates. In vivo, most of these systems help their native organism to react to environmental clues by regulating expression of for instance catabolic enzymes or importers.
In the past, these have ben co-opted by researchers to function as sensors or to control enzyme expression to regulate a pathway depending on the availability of a certain metabolite
(Rogers, Taylor, and Church 2016).
By using reporter genes like GFP or the LUX operon, we have a measurable output that can be used to select for the best contenders. By running this in several iterations, nature's most powerful optimization tool, evolution, can be applied to further improve the pathway
(Williams et al. 2016).
Applying fluorescence activated cell sorting (FACS) coupled with
V. natriegens
incredible doubling time, thousands of generations can be screened for progressively better pathway variants.
So, we decided to put our toolbox full of characterized parts to good use. The Marburg collection contains all the parts needed to find the ideal enzyme balance for highest production. Being able to build a large number of differentially assembled pathways is the answer to creating the most productive strain. Finding the right one in that complexity is another difficulty to overcome, one we solved by finding transcriptional regulators that specifically activate reporter gene expression relative to the concentration of key metabolites, 3-hydroxypropionate and Malonyl-CoA. This gives us a quantifiable output with which we are able to judge the different combinations. Also, utilizing the speed of V. natriegens , the time required for a high throughput screening can be reduced dramatically.
We deemed the concentration of two different metabolites as being critical to our project: 3-Hydroxypropionate, the product itself, and malonyl-CoA, an important intermediate we also plan to overexpress.

Using the information from the paper of Hanko et al. (2017), we settled for the hdpR / hdpH system coming from Pseudomonas putida KT2440. It consists the constitutively expressed HdpR protein (BBa_K2560304). , which is able to bind to the P HdpR and induce expression of a gene. In the native setting, that would be the hdpH gene, encoding for a 3-hydroxypropionate dehydrogenase which enables P. putida to grow on 3HPA . We cloned our reporter gene in its position. This way we got a direct readout of the 3HPA concentrations in the individual cells.
Because our model showed that the low activity of
V. natriegens
native Acc , malonyl-CoA is likely to be a limiting factor in 3HPA production. Having a direct indication to how much malonyl-CoA is available

Directed evolution
 Although the Marburg Collection enables the construction of many pathway variations, there are still many more possibilities for direct metabolic fluxes towards 3HPA . To enforce these flux improvements, we planned to use directed evolution coupled with FACS selection.
By implementing genes for the conversion of 3HPA into succinate, we lead our product back to the central metabolism. By deleting all other genes responsible for succinate production, the cells are forced to use the 3HPA bypass to get succinate, which is needed in the TCA cycle.
The big advantage of using V. natriegens for that purpose is that it’s short doubling time increases the number of cell divisions per time and thereby increases the number of mutations in a given time frame.
Coupled with our biosensors and fluorescence-activated cell sorting (FACS) we can easily and rapidly select for cells performing the most efficient pathways.
Additionally to mutation-guided evolution there are many genome modifications which could increase flow towards 3HPA , for instance by downregulation of side reactions. In the Directed Evolution section of our metabolic engineering design, a list of possible genome modifications is shown.
By using MAGE , it is possible to get a library of different genome modifications, which can then be selected via FACS for their efficiency to produce 3HPA or succinate. MAGE stands for multiplex automated genome engineering and is a technique to create combinatorial genome modifications.
MAGE uses a set of oligos, which get transformed into a population of cells and then integrate into the chromosome via homologous recombination.
Different cells in the population will have different numbers and combinations of modifications leading to a genomic diversity. By applying MAGE in combination with FACS for our pathway we get a huge variety of possible pathway optimizations which can be sorted via FACS based on their efficiency.
Although the Marburg Collection enables the construction of many pathway variations, there are still many more possibilities for direct metabolic fluxes towards 3HPA . To enforce these flux improvements, we planned to use directed evolution coupled with FACS selection.
By implementing genes for the conversion of 3HPA into succinate, we lead our product back to the central metabolism. By deleting all other genes responsible for succinate production, the cells are forced to use the 3HPA bypass to get succinate, which is needed in the TCA cycle.
The big advantage of using V. natriegens for that purpose is that it’s short doubling time increases the number of cell divisions per time and thereby increases the number of mutations in a given time frame.
Coupled with our biosensors and fluorescence-activated cell sorting (FACS) we can easily and rapidly select for cells performing the most efficient pathways.
Additionally to mutation-guided evolution there are many genome modifications which could increase flow towards 3HPA , for instance by downregulation of side reactions. In the Directed Evolution section of our metabolic engineering design, a list of possible genome modifications is shown.
By using MAGE , it is possible to get a library of different genome modifications, which can then be selected via FACS for their efficiency to produce 3HPA or succinate. MAGE stands for multiplex automated genome engineering and is a technique to create combinatorial genome modifications.
MAGE uses a set of oligos, which get transformed into a population of cells and then integrate into the chromosome via homologous recombination.
Different cells in the population will have different numbers and combinations of modifications leading to a genomic diversity. By applying MAGE in combination with FACS for our pathway we get a huge variety of possible pathway optimizations which can be sorted via FACS based on their efficiency.
Applications of our workflow
By using V. natriegens as a chassis for metabolic engineering, it is possible to build, test and optimize pathways and enzymes at a high-throughput level. The Marburg Collection enables the fast construction of pathway libraries, while the genetic accessibility of the strain makes genome modification and pathway integration simple. The usage of our biosensors for screening of malonyl-CoA and 3HPA accelerates identification of the best pathway variants and the short doubling time coupled with the high nutrient uptake rate empowers V. natriegens to be the perfect chassis for metabolic and enzyme engineering!
Aiyar, Sarah E, Tamas Gaal, and Richard L Gourse. 2002. RRNA Promoter Activity in the Fast-Growing Bacterium Vibrio Natriegens. Journal of bacteriology 184(5): 1349-58. http://www.ncbi.nlm.nih.gov/pubmed/11844764 (August 23, 2018).
Austin, Payne B, A Zachary, and R R Colwell. 2018. 28 International Association of Microbiological Societies Recognition of Beneckea Natriegens (Payne et al.; Baumann et al.) as a Member of the Genus Vibrio, as Previously Proposed by Webb and Payne . www.microbFiologyresearch.org (August 23, 2018).
Bi, Keran et al. 2016. Isolation and Molecular Identification of Vibrio Natriegens from Diseased Portunus Trituberculatus in China. Journal of the World Aquaculture Society 47(6): 854-61. http://doi.wiley.com/10.1111/jwas.12305 (September 29, 2018).
Carlson, Erik D, Rui Gan, C Eric Hodgman, and Michael C Jewett. 2012. Cell-Free Protein Synthesis: Applications Come of Age. Biotechnology advances 30(5): 1185–94. http://www.ncbi.nlm.nih.gov/pubmed/22008973 (October 1, 2018).
CHIEN, C et al. 2007. “Production of Poly-β-Hydroxybutyrate (PHB) by Vibrio Spp. Isolated from Marine Environment.” Journal of Biotechnology 132(3): 259–63. http://www.ncbi.nlm.nih.gov/pubmed/17416432 (September 22, 2018).
Coyer, J A, A Cabello-Pasini, H Swift, and R S Alberte. 1996. “N2 Fixation in Marine Heterotrophic Bacteria: Dynamics of Environmental and Molecular Regulation.” Proceedings of the National Academy of Sciences of the United States of America 93(8): 3575–80. http://www.ncbi.nlm.nih.gov/pubmed/11607653 (August 23, 2018).
Dalia, Triana N. et al. 2017. “Multiplex Genome Editing by Natural Transformation (MuGENT) for Synthetic Biology in Vibrio Natriegens .” ACS Synthetic Biology 6(9): 1650–55. http://www.ncbi.nlm.nih.gov/pubmed/28571309 (September 30, 2018).
Delpech, Roger. 2001. “Using Vibrio Natriegetis for Studying Bacterial Population Growth, Artificial Selection, and the Effects of UV Radiation and Photo-Reactivation.” Journal of Biological Education 35(2): 93–97. http://www.tandfonline.com/doi/abs/10.1080/00219266.2000.9655749 (August 23, 2018).
EAGON, R G. 1962. “Pseudomonas Natriegens, a Marine Bacterium with a Generation Time of Less than 10 Minutes.” Journal of bacteriology 83(4): 736–37. http://www.ncbi.nlm.nih.gov/pubmed/13888946 (August 23, 2018).
Failmezger, Jurek, Steffen Scholz, Bastian Blombach, and Martin Siemann-Herzberg. 2018. “Cell-Free Protein Synthesis From Fast-Growing Vibrio Natriegens.” Frontiers in Microbiology 9: 1146. https://www.frontiersin.org/article/10.3389/fmicb.2018.01146/full (September 30, 2018).
Fernández-Llamosas, Helga et al. 2017. “Speeding up Bioproduction of Selenium Nanoparticles by Using Vibrio Natriegens as Microbial Factory.” Scientific Reports 7(1): 16046. http://www.nature.com/articles/s41598-017-16252-1 (September 28, 2018).
Hoffart, Eugenia et al. 2017. “High Substrate Uptake Rates Empower Vibrio Natriegens as Production Host for Industrial Biotechnology.” Applied and environmental microbiology : AEM.01614-17. http://www.ncbi.nlm.nih.gov/pubmed/28887417 (October 6, 2018).
Lee, Henry H et al. 2016. “Vibrio Natriegens, a New Genomic Powerhouse.” bioRxiv : 058487. https://www.biorxiv.org/content/early/2016/06/12/058487 (September 30, 2018).
Lee, Henry H, Nili Ostrov, Michaela A Gold, and George M Church. 2017. “Recombineering in Vibrio Natriegens.” bioRxiv : 130088. https://www.biorxiv.org/content/early/2017/04/26/130088 (September 30, 2018).
Long, Christopher P., Jacqueline E. Gonzalez, Robert M. Cipolla, and Maciek R. Antoniewicz. 2017. “Metabolism of the Fast-Growing Bacterium Vibrio Natriegens Elucidated by 13C Metabolic Flux Analysis.” Metabolic Engineering 44: 191–97. http://www.ncbi.nlm.nih.gov/pubmed/29042298 (October 5, 2018).
Maida, I. et al. 2013. “Draft Genome Sequence of the Fast-Growing Bacterium Vibrio Natriegens Strain DSMZ 759.” Genome Announcements 1(4). http://www.ncbi.nlm.nih.gov/pubmed/23969053 (August 23, 2018).
Mehbub, Mohammad F. et al. 2018. “A Controlled Aquarium System and Approach to Study the Role of Sponge-Bacteria Interactions Using Aplysilla Rosea and Vibrio Natriegens.” Scientific Reports 8(1): 11801. http://www.nature.com/articles/s41598-018-30295-y (September 29, 2018).
Miura, A et al. 1981. “Growth-Rate-Dependent Regulation of Ribosome Synthesis in E. coli: Expression of the LacZ and GalK Genes Fused to Ribosomal Promoters.” Cell 25(3): 773–82. http://www.ncbi.nlm.nih.gov/pubmed/6793240 (September 22, 2018).
Mullenger, L., and Nijole R. Gill. 1973. “Vibrio Natriegens: A Rapidly Growing Micro-Organism Ideally Suited for Class Experiments.” Journal of Biological Education 7(5): 33–39. http://www.tandfonline.com/doi/abs/10.1080/00219266.1973.9653881 (August 21, 2018).
Nazly, E D. 1980. 116 Journal of General Microbiology Adenine Nucleotide Pools During Starvation of Beneckea Natviegens . www.microbiologyresearch.org (October 7, 2018).
Nissen, Hilde, Mikal Heldal, and Svein Norland. 1987. “Growth, Elemental Composition, and Formation of Polyphosphate Bodies in Vibrio Natriegens Cultures Shifted from Phosphate-Limited to Phosphate-Pulsed Media.” Canadian Journal of Microbiology 33(7): 583–88. http://www.nrcresearchpress.com/doi/10.1139/m87-101 (September 22, 2018).
Payne, W J, R G Eagon, and A K Williams. 1960. SOME OBSERVATIONS ON THE PHYSIOLOGY OF PSEUDOMONAS NATRIEGENS NOV. SPEC. 1) . https://link.springer.com/content/pdf/10.1007/BF02538432.pdf (August 21, 2018).
Payne, William J. STUDIES ON BACTERIAL UTILIZATION OF URONIC ACIDS III. INDUCTION OF OXIDATIVE ENZYMES IN A MARINE ISOLATE’ . https://www.ncbi.nlm.nih.gov/pmc/articles/PMC290205/pdf/jbacter00506-0093.pdf (August 21, 2018).
Pollack-Berti, Amber, Michael S. Wollenberg, and Edward G. Ruby. 2010. “Natural Transformation of Vibrio Fischeri Requires TfoX and TfoY.” Environmental Microbiology 12(8): no-no. http://doi.wiley.com/10.1111/j.1462-2920.2010.02250.x (September 30, 2018).
Simons, Keryn L., Susan M. Thomas, and Peter A. Anderson. 2010. “Identification of Vibrio Natriegens UvrA and UvrB Genes and Analysis of Gene Regulation Using Transcriptional Reporter Plasmids.” The Journal of Microbiology 48(5): 644–56. http://www.ncbi.nlm.nih.gov/pubmed/21046343 (September 22, 2018).
Des Soye, Benjamin J. et al. 2018. “Establishing a High-Yielding Cell-Free Protein Synthesis Platform Derived from Vibrio Natriegens .” ACS Synthetic Biology 7(9): 2245–55. http://www.ncbi.nlm.nih.gov/pubmed/30107122 (September 30, 2018).
Webb, C D, and W J Payne. 1971. “Influence of Na+ on Synthesis of Macromolecules by a Marine Bacterium.” Applied microbiology 21(6): 1080–88. http://www.ncbi.nlm.nih.gov/pubmed/4327612 (August 23, 2018).
Weinstock, Matthew T, Eric D Hesek, Christopher M Wilson, and Daniel G Gibson. 2016. “Vibrio Natriegens as a Fast-Growing Host for Molecular Biology.” Nature Methods 13(10): 849–51. http://www.ncbi.nlm.nih.gov/pubmed/27571549 (September 29, 2018).
Wiegand, Daniel J., Henry H. Lee, Nili Ostrov, and George M. Church. 2018. “Establishing a Cell-Free Vibrio Natriegens Expression System.” ACS Synthetic Biology : acssynbio.8b00222. http://pubs.acs.org/doi/10.1021/acssynbio.8b00222 (September 30, 2018).
Zachary, A. 1976. “Physiology and Ecology of Bacteriophages of the Marine Bacterium Beneckea Natriegens: Salinity.” Applied and environmental microbiology 31(3): 415–22. http://www.ncbi.nlm.nih.gov/pubmed/938035 (August 23, 2018).
Zachary, Arthur. 1974. 27 APPLIED MICROBIOLOGY NOTES Isolation of Bacteriophages of the Marine Bacterium Beneckea Natriegens from Coastal Salt Marshes’ . https://www.ncbi.nlm.nih.gov/pmc/articles/PMC380186/pdf/applmicro00040-0188.pdf (September 28, 2018).
Zacheray, A. 1978. An Ecological Study of Bacteriophages of Vibrio Natriegens . Canadian Journal of Microbiology 24(3): 321-24. http://www.nrcresearchpress.com/doi/10.1139/m78-053 (September 23, 2018).
Brazelton, VA et al. 2016. A quick guide to CRISPR sgRNA design tools. GM Crops Food . 2015; 6(4): 266–276. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5033207/pdf/kgmc-06-04-1137690.pdf
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S., Dohmae, N., Ishitani, R., Zhang, F., Nureki, O. (2014) Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA, Cell , 156 (5), 935-949. https://www.cell.com/action/showPdf?pii=S0092-8674%2814%2900156-1
Jinek, M., Jiang, F., Taylor, D. W., Sternberg, S. H., Kaya, E., Ma, E., Anders, C., Hauer, M., Zhou, K., Lin, S., Kaplan, M., Iavarone, A. T., Charpentier, E., Nogale, E., Doudna, J. A. (2014) Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation, Science , 343 (6176), 1-28. http://science.sciencemag.org/content/343/6176/1247997
Deltcheva, E., Chylinski, K., Sharma, C. M., Gonzales, K., Chao, Y., Pirzada, Z. A., Eckert, M. R., Vogel, J., Charpentier, E. (2011) CRISPR RNA maturation by trans -encoded small RNA and host factor RNase III, Nature , 471, 602-609. https://www.nature.com/articles/nature09886
Gasiunas, R. Barrangou, P. Horvath, V. Siksnys. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria.
Proc. Natl. Acad. Sci. USA , 109 (2012), pp. E2579-E2586. http://www.pnas.org/content/pnas/109/39/E2579.full.pdf
Jinek, K. Chylinski, I. Fonfara, M. Hauer, J.A. Doudna, E. Charpentier. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science , 337 (2012), pp. 816-821. http://science.sciencemag.org/content/337/6096/816
Doudna, J. A. & Charpentier, E. 2014.Genome editing. The new frontier of genome engineering with CRISPR-Cas9 . Science 346, 1258096 (2014). http://science.sciencemag.org/content/346/6213/1258096.full
Charpentier, E. & Marraffini, L. A. 2014. Harnessing CRISPR-Cas9 immunity for genetic engineering . Curr. Opin. Microbiol. 19, 114–119 (2014). https://www.sciencedirect.com/science/article/pii/S1369527414000885?via%3Dihub
Hsu, P. D., Lander, E. S., Zhang, F. (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering, Cell , 157, 1262-1276. https://www.cell.com/action/showPdf?pii=S0092-8674%2814%2900604-7
Mazhar Adli. 2018. The CRISPR tool kit for genome editing and beyond. Nature Communications volume 9, Article number: 1911 (2018) https://www.nature.com/articles/s41467-018-04252-2.pdf
Jansen, R., van Embden, J. D. A., Gaastra, W., Schouls, L. M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes, Molecular Microbiology , 43 (6), 1565-1575. https://onlinelibrary.wiley.com/doi/epdf/10.1046/j.1365-2958.2002.02839.x
Ishino, Y., Shinagawa, H., Makino, K., Amemura, M., Nakata, A. (1987) Nucleotide Sequence of the iap Gene, Responsible for Alkaline Phosphatase Isozyme Conversion in Escherichia coli , and Identification of the Gene Product, Journal of Bacteriology , 169 (12), 5429-5433. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC213968/pdf/jbacter00202-0107.pdf
Qi et al. 2013. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013 Feb 28; 152(5): 1173–1183. https://www.cell.com/action/showPdf?pii=S0092-8674%2813%2900211-0
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A., Horvath, P. (2007) CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes,
Science
, 315, 1709-1712. http://science.sciencemag.org/content/315/5819/1709
Brazelton, VA et al. 2016. A quick guide to CRISPR sgRNA design tools. GM Crops Food . 2015; 6(4): 266–276. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5033207/pdf/kgmc-06-04-1137690.pdf
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S., Dohmae, N., Ishitani, R., Zhang, F., Nureki, O. (2014) Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA, Cell , 156 (5), 935-949. https://www.cell.com/action/showPdf?pii=S0092-8674%2814%2900156-1
Jinek, M., Jiang, F., Taylor, D. W., Sternberg, S. H., Kaya, E., Ma, E., Anders, C., Hauer, M., Zhou, K., Lin, S., Kaplan, M., Iavarone, A. T., Charpentier, E., Nogale, E., Doudna, J. A. (2014) Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation, Science , 343 (6176), 1-28. http://science.sciencemag.org/content/343/6176/1247997
Deltcheva, E., Chylinski, K., Sharma, C. M., Gonzales, K., Chao, Y., Pirzada, Z. A., Eckert, M. R., Vogel, J., Charpentier, E. (2011) CRISPR RNA maturation by trans -encoded small RNA and host factor RNase III, Nature , 471, 602-609. https://www.nature.com/articles/nature09886
Proc. Natl. Acad. Sci. USA , 109 (2012), pp. E2579-E2586. http://www.pnas.org/content/pnas/109/39/E2579.full.pdf
Jinek, K. Chylinski, I. Fonfara, M. Hauer, J.A. Doudna, E. Charpentier. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science , 337 (2012), pp. 816-821. http://science.sciencemag.org/content/337/6096/816
Doudna, J. A. & Charpentier, E. 2014.Genome editing. The new frontier of genome engineering with CRISPR-Cas9 . Science 346, 1258096 (2014). http://science.sciencemag.org/content/346/6213/1258096.full
Charpentier, E. & Marraffini, L. A. 2014. Harnessing CRISPR-Cas9 immunity for genetic engineering . Curr. Opin. Microbiol. 19, 114–119 (2014). https://www.sciencedirect.com/science/article/pii/S1369527414000885?via%3Dihub
Hsu, P. D., Lander, E. S., Zhang, F. (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering, Cell , 157, 1262-1276. https://www.cell.com/action/showPdf?pii=S0092-8674%2814%2900604-7
Mazhar Adli. 2018. The CRISPR tool kit for genome editing and beyond. Nature Communications volume 9, Article number: 1911 (2018) https://www.nature.com/articles/s41467-018-04252-2.pdf
Jansen, R., van Embden, J. D. A., Gaastra, W., Schouls, L. M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes, Molecular Microbiology , 43 (6), 1565-1575. https://onlinelibrary.wiley.com/doi/epdf/10.1046/j.1365-2958.2002.02839.x
Ishino, Y., Shinagawa, H., Makino, K., Amemura, M., Nakata, A. (1987) Nucleotide Sequence of the iap Gene, Responsible for Alkaline Phosphatase Isozyme Conversion in Escherichia coli , and Identification of the Gene Product, Journal of Bacteriology , 169 (12), 5429-5433. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC213968/pdf/jbacter00202-0107.pdf
Qi et al. 2013. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013 Feb 28; 152(5): 1173–1183. https://www.cell.com/action/showPdf?pii=S0092-8674%2813%2900211-0
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A., Horvath, P. (2007) CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes,
Science
, 315, 1709-1712. http://science.sciencemag.org/content/315/5819/1709
Alber, B.E., Fuchs, G., 2002. Propionyl-coenzyme a synthase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Biol. Chem. 277, 12137–12143.
Gande, R., Dover, L.G., Krumbach, K., Besra, G.S., Sahm, H., Oikawa, T., Eggeling, L., 2007. The two carboxylases of corynebacterium glutamicum essential for fatty acid and mycolic acid synthesis. J. Bacteriol. 189, 5257–5264.
Hanko, E.K.R., Minton, N.P., Malys, N., 2017. Characterisation of a 3-hydroxypropionic acid-inducible system from Pseudomonas putida for orthogonal gene expression control in Escherichia coli and Cupriavidus necator. Sci. Rep. 7, 1724.
Janßen, H.J., Steinbüchel, A., 2014. Fatty acid synthesis in Escherichia coli and its applications towards the production of fatty acid based biofuels. Biotechnol Biofuels 7, 7.
Kildegaard, K.R., Jensen, N.B., Schneider, K., Czarnotta, E., Özdemir, E., Klein, T., Maury, J., Ebert, B.E., Christensen, H.B., Chen, Y., Kim, I.K., Herrgård, M.J., Blank, L.M., Forster, J., Nielsen, J., Borodina, I., 2016. Engineering and systems-level analysis of Saccharomyces cerevisiae for production of 3-hydroxypropionic acid via malonyl-CoA reductase-dependent pathway. Microb. Cell Fact. 15.
Lee, C.-K., Cheong, H.-K., Ryu, K.-S., Lee, J. Il, Lee, W., Jeon, Y.H., Cheong, C., 2008. Biotinoyl domain of human acetyl-CoA carboxylase: Structural insights into the carboxyl transfer mechanism. Proteins Struct. Funct. Bioinforma. 72, 613–624.
Liu, C., Ding, Y., Xian, M., Liu, M., Liu, H., Ma, Q., Zhao, G., 2017. Malonyl-CoA pathway: a promising route for 3-hydroxypropionate biosynthesis. Crit. Rev. Biotechnol. 37, 933–941.
Liu, C., Wang, Q., Xian, M., Ding, Y., Zhao, G., 2013. Dissection of Malonyl-Coenzyme A Reductase of Chloroflexus aurantiacus Results in Enzyme Activity Improvement. PLoS One 8, 1–8.
Raj, S.M., Rathnasingh, C., Jo, J.E., Park, S., 2008. Production of 3-hydroxypropionic acid from glycerol by a novel recombinant Escherichia coli BL21 strain. Process Biochem. 43, 1440–1446.
Rathnasingh, C., Raj, S.M., Lee, Y., Catherine, C., Ashok, S., Park, S., 2012. Production of 3-hydroxypropionic acid via malonyl-CoA pathway using recombinant Escherichia coli strains. J. Biotechnol. 157, 633–640.
Ro, D.K., Paradise, E.M., Quellet, M., Fisher, K.J., Newman, K.L., Ndungu, J.M., Ho, K.A., Eachus, R.A., Ham, T.S., Kirby, J., Chang, M.C.Y., Withers, S.T., Shiba, Y., Sarpong, R., Keasling, J.D., 2006. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440, 940–943.
Stephanopoulos, G., 2012. Synthetic biology and metabolic engineering. ACS Synth. Biol. 1, 514–525.
Stephanopoulos, G., 2007. Challenges in engineering microbes for biofuels production. Science 315, 801–4.
Valdehuesa, K.N.G., Liu, H., Nisola, G.M., Chung, W.J., Lee, S.H., Park, S.J., 2013. Recent advances in the metabolic engineering of microorganisms for the production of 3-hydroxypropionic acid as C3 platform chemical. Appl. Microbiol. Biotechnol. 97, 3309–3321.
Vidra, A., Németh, Á., 2017. Bio-based 3-hydroxypropionic Acid : A Review. period. Polytech. Chem. Eng. 1–11.
Werpy, T., Petersen, G., 2004. Top Value Added Chemicals from Biomass Volume I — Results of Screening for Potential Candidates from Sugars and Synthesis Gas Top Value Added Chemicals From Biomass Volume I : Results of Screening for Potential Candidates. Other Inf. PBD 1 Aug 2004 Medium: ED; Size: 76 pp. pages.
Antonova ES, Hammer BK. Genetics of Natural Competence in Vibrio cholerae and other Vibrios. Microbiol Spectr. (2015);3(3).
Argos, P., A. Landy, K. Abremski, J. B. Egan, E. Haggard-Ljungquist, R. H. Hoess, M. L. Kahn, B. Kalionis, S. V. Narayana, L. S. Pierson, 3rd and et al. (1986). "The integrase family of site-specific recombinases: regional similarities and global diversity." EMBO J 5(2): 433-440.
Blokesch, M. (2012). "TransFLP--a method to genetically modify Vibrio cholerae based on natural transformation and FLP-recombination." J Vis Exp(68).
Blokesch, M., Schoolnik, G. K., The Extracellular Nuclease Dns and Its Role in Natural Transformation of Vibrio cholerae, JOURNAL OF BACTERIOLOGY, (2008), 7232–7240.
Broach, J. R., V. R. Guarascio and M. Jayaram (1982). "Recombination within the yeast plasmid 2mu circle is site-specific." Cell 29(1): 227-234.
Bryan, FR., Construction of a recombinase-deficient mutant recA protein that retains single-stranded DNA-dependent ATPase activity, J Biol Chem. (1988); 263(18):8716-23.
Burn, S.F., Detection of β-galactosidase activity: X-gal staining. Methods Mol Biol. 2012;886:241-50.
Chen, Z., Yang, H., Pavletich N. P., Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures, Nature (2008) 22;453(7194):489-4.
Cheetham, G. M. & Steitz, T. A. Structure of a transcribing T7 RNA polymerase initiation complex. Science (New York, N.Y.) 286, 2305–2309 (1999).
Chung, C. H. and A. L. Goldberg (1981). "The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La." Proc Natl Acad Sci U S A 78(8): 4931-4935.
Dalia, T. N., C. A. Hayes, S. Stolyar, C. J. Marx, J. B. McKinlay and A. B. Dalia (2017). "Multiplex Genome Editing by Natural Transformation (MuGENT) for Synthetic Biology in Vibrio natriegens." ACS Synth Biol 6(9): 1650-1655.
Donnenberg, M. S. and J. B. Kaper (1991). "Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector." Infect Immun 59(12): 4310-4317.
Evans, B. R., J. W. Chen, R. L. Parsons, T. K. Bauer, D. B. Teplow and M. Jayaram (1990). "Identification of the active site tyrosine of Flp recombinase. Possible relevance of its location to the mechanism of recombination." J Biol Chem 265(30): 18504-18510.
Fuchs, G., Allgemeine Mikrobiologie, Thieme. (2017).
Fields, S. & O. Song, (1989) A novel genetic system to detect protein-protein interactions. Nature 340: 245-246.https://www.ncbi.nlm.nih.gov/pubmed/2547163
Karimova, G., J. Pidoux, A. Ullmann & D. Ladant, (1998) A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proceedings of the National Academy of Sciences of the United States of America 95: 5752-5756.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC20451/
Kolb, A., S. Busby, H. Buc, S. Garges & S. Adhya, (1993) Transcriptional regulation by cAMP and its receptor protein. Annual review of biochemistry 62: 749-795.
https://www.ncbi.nlm.nih.gov/pubmed/8394684
Krupinski, J., (1991) The adenylyl cyclase family. Molecular and cellular biochemistry 104: 73-79. https://link.springer.com/article/10.1007/BF00229806
Griffith F, The significance of pneumococcal types. Journal of Hygiene. 27, Nr. 2. (1928) S. 113–59.
Gill, R. T., M. P. DeLisa, M. Shiloach, T. R. Holoman and W. E. Bentley (2000). "OmpT expression and activity increase in response to recombinant chloramphenicol acetyltransferase overexpression and heat shock in E. coli." J Mol Microbiol Biotechnol 2(3): 283-289.
Grodberg, J. and J. J. Dunn (1988). "ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification." J Bacteriol 170(3): 1245-1253.
Horii T.; Ogawa T.; Nakatani T.; Hase T.; Matsubara H. & Ogawa H., Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein, cell (1981) Vol. 27, 515-522.
Hartmann, G., Honikel, K. O., Knüsel, F. & Nüesch, J. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochimica et biophysica acta 145, 843–844 (1967).
Howard-Flanders, P., E. Simson and L. Theriot (1964). "A Locus That Controls Filament Formation and Sensitivity to Radiation in Escherichia Coli K-12." Genetics 49: 237-246.
Huisman, O., R. D'Ari and S. Gottesman (1984). "Cell-division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation." Proc Natl Acad Sci U S A 81(14): 4490-4494.
Jeong, H., Kim, H. J. & Lee, S. J. Complete Genome Sequence of Escherichia coli Strain BL21. Genome announcements 3 (2015).
Lorenz, M. G., and Wackernagel, W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. (1994) 58, 563−602.
Langley, KE., Villarejo, M.R., Fowler, A.V., Zamenhof, P.J., and Zabin, I, Molecular basis of beta-galactosidase alpha-complementation. Proc Natl Acad Sci U S A. (1975);72(4):1254-7.
Leytus, S. P., L. K. Bowles, J. Konisky and W. F. Mangel (1981). "Activation of plasminogen to plasmin by a protease associated with the outer membrane of Escherichia coli." Proc Natl Acad Sci U S A 78(3): 1485-1489.
Lundrigan, M. D. and R. M. Webb (1992). "Prevalence of ompT among Escherichia coli isolates of human origin." FEMS Microbiol Lett 76(1-2): 51-56.
Meibom, K. L., M. Blokesch, N. A. Dolganov, C. Y. Wu and G. K. Schoolnik (2005). "Chitin induces natural competence in Vibrio cholerae." Science 310(5755): 1824-1827.
Mell JC, Redfield RJ. Natural competence and the evolution of DNA uptake specificity. J Bacteriol. (2014);196(8):1471-83.
Mizusawa, S. and S. Gottesman (1983). "Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein." Proc Natl Acad Sci U S A 80(2): 358-362.
Nishino, T., Morikawa, T., Structure and function of nucleases in DNA repair: shape, grip and blade of the DNA scissors, Oncogene (2002) 21, 9022 – 9032.
Pan, G. and P. D. Sadowski (1992). "Ligation activity of FLP recombinase. The strand ligation activity of a site-specific recombinase using an activated DNA substrate." J Biol Chem 267(18): 12397-12399.
Pollack-Berti, A., M. S. Wollenberg and E. G. Ruby (2010). "Natural transformation of Vibrio fischeri requires tfoX and tfoY." Environ Microbiol 12(8): 2302-2311.
Sadowski, P. D. (1995). "The Flp recombinase of the 2-microns plasmid of Saccharomyces cerevisiae." Prog Nucleic Acid Res Mol Biol 51: 53-91.
Susan T. Lovett, The DNA exonucleases of Escherichia coli, (2003) 4(2).
Savir, Y.,Tlusty, T., RecA-mediated homology search as a nearly optimal signal detection system, Molecular Cell (2010) 40, 388–396.
Santillán M, Mackey MC., Quantitative approaches to the study of bistability in the lac operon of Escherichia coli., J R Soc Interface. (2008) 6;5 Suppl 1:S29-39.
Serebriiskii I.G., Golemis E.A., Uses of lacZ to study gene function: evaluation of β-galactosidase assays employed in the yeast two-hybrid system. Anal. Biochem. 2000;285:1–15.
Studier, F. W. & Moffatt, B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of molecular biology 189, 113–130 (1986).
Sodeinde, O. A., Y. V. Subrahmanyam, K. Stark, T. Quan, Y. Bao and J. D. Goguen (1992). "A surface protease and the invasive character of plague." Science 258(5084): 1004-1007.
Tabor, S. & Richardson, C. C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proceedings of the National Academy of Sciences of the United States of America 82, 1074–1078 (1985).
Villaverde, A. and M. M. Carrio (2003). "Protein aggregation in recombinant bacteria: biological role of inclusion bodies." Biotechnol Lett 25(17): 1385-1395.
Wu R., Zhao M., Li J., Gao H., Kan B., Liang W. Direct regulation of the natural competence regulator gene tfoX by cyclic AMP (cAMP) and cAMP receptor protein (CRP) in Vibrios. Sci Rep. (2015) 7;5:14921.
Weinstock, M. T., E. D. Hesek, C. M. Wilson and D. G. Gibson (2016). "Vibrio natriegens as a fast-growing host for molecular biology." Nat Methods 13(10): 849-851.
Zamenhof, P.J., Villarejo, M. Construction and Properties of E. coli Strains Exhibiting α-Complementation of β-Galactosidase Fragments In Vivo. J Bacteriol. (1972); 110(1): 171–178.
Antonova ES, Hammer BK. Genetics of Natural Competence in Vibrio cholerae and other Vibrios. Microbiol Spectr. (2015);3(3).
Argos, P., A. Landy, K. Abremski, J. B. Egan, E. Haggard-Ljungquist, R. H. Hoess, M. L. Kahn, B. Kalionis, S. V. Narayana, L. S. Pierson, 3rd and et al. (1986). "The integrase family of site-specific recombinases: regional similarities and global diversity." EMBO J 5(2): 433-440.
Blokesch, M. (2012). "TransFLP--a method to genetically modify Vibrio cholerae based on natural transformation and FLP-recombination." J Vis Exp(68).
Blokesch, M., Schoolnik, G. K., The Extracellular Nuclease Dns and Its Role in Natural Transformation of Vibrio cholerae, JOURNAL OF BACTERIOLOGY, (2008), 7232–7240.
Broach, J. R., V. R. Guarascio and M. Jayaram (1982). "Recombination within the yeast plasmid 2mu circle is site-specific." Cell 29(1): 227-234.
Bryan, FR., Construction of a recombinase-deficient mutant recA protein that retains single-stranded DNA-dependent ATPase activity, J Biol Chem. (1988); 263(18):8716-23.
Burn, S.F., Detection of β-galactosidase activity: X-gal staining. Methods Mol Biol. 2012;886:241-50.
Chen, Z., Yang, H., Pavletich N. P., Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures, Nature (2008) 22;453(7194):489-4.
Cheetham, G. M. & Steitz, T. A. Structure of a transcribing T7 RNA polymerase initiation complex. Science (New York, N.Y.) 286, 2305–2309 (1999).
Chung, C. H. and A. L. Goldberg (1981). "The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La." Proc Natl Acad Sci U S A 78(8): 4931-4935.
Dalia, T. N., C. A. Hayes, S. Stolyar, C. J. Marx, J. B. McKinlay and A. B. Dalia (2017). "Multiplex Genome Editing by Natural Transformation (MuGENT) for Synthetic Biology in Vibrio natriegens." ACS Synth Biol 6(9): 1650-1655.
Donnenberg, M. S. and J. B. Kaper (1991). "Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector." Infect Immun 59(12): 4310-4317.
Evans, B. R., J. W. Chen, R. L. Parsons, T. K. Bauer, D. B. Teplow and M. Jayaram (1990). "Identification of the active site tyrosine of Flp recombinase. Possible relevance of its location to the mechanism of recombination." J Biol Chem 265(30): 18504-18510.
Fuchs, G., Allgemeine Mikrobiologie, Thieme. (2017).
Fields, S. & O. Song, (1989) A novel genetic system to detect protein-protein interactions. Nature 340: 245-246.https://www.ncbi.nlm.nih.gov/pubmed/2547163
Karimova, G., J. Pidoux, A. Ullmann & D. Ladant, (1998) A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proceedings of the National Academy of Sciences of the United States of America 95: 5752-5756.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC20451/
Kolb, A., S. Busby, H. Buc, S. Garges & S. Adhya, (1993) Transcriptional regulation by cAMP and its receptor protein. Annual review of biochemistry 62: 749-795.
https://www.ncbi.nlm.nih.gov/pubmed/8394684
Krupinski, J., (1991) The adenylyl cyclase family. Molecular and cellular biochemistry 104: 73-79. https://link.springer.com/article/10.1007/BF00229806
Griffith F, The significance of pneumococcal types. Journal of Hygiene. 27, Nr. 2. (1928) S. 113–59.
Gill, R. T., M. P. DeLisa, M. Shiloach, T. R. Holoman and W. E. Bentley (2000). "OmpT expression and activity increase in response to recombinant chloramphenicol acetyltransferase overexpression and heat shock in E. coli." J Mol Microbiol Biotechnol 2(3): 283-289.
Grodberg, J. and J. J. Dunn (1988). "ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification." J Bacteriol 170(3): 1245-1253.
Horii T.; Ogawa T.; Nakatani T.; Hase T.; Matsubara H. & Ogawa H., Regulation of SOS functions: Purification of E. coli LexA protein and determination of its specific site cleaved by the RecA protein, cell (1981) Vol. 27, 515-522.
Hartmann, G., Honikel, K. O., Knüsel, F. & Nüesch, J. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochimica et biophysica acta 145, 843–844 (1967).
Howard-Flanders, P., E. Simson and L. Theriot (1964). "A Locus That Controls Filament Formation and Sensitivity to Radiation in Escherichia Coli K-12." Genetics 49: 237-246.
Huisman, O., R. D'Ari and S. Gottesman (1984). "Cell-division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation." Proc Natl Acad Sci U S A 81(14): 4490-4494.
Jeong, H., Kim, H. J. & Lee, S. J. Complete Genome Sequence of Escherichia coli Strain BL21. Genome announcements 3 (2015).
Lorenz, M. G., and Wackernagel, W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. (1994) 58, 563−602.
Langley, KE., Villarejo, M.R., Fowler, A.V., Zamenhof, P.J., and Zabin, I, Molecular basis of beta-galactosidase alpha-complementation. Proc Natl Acad Sci U S A. (1975);72(4):1254-7.
Leytus, S. P., L. K. Bowles, J. Konisky and W. F. Mangel (1981). "Activation of plasminogen to plasmin by a protease associated with the outer membrane of Escherichia coli." Proc Natl Acad Sci U S A 78(3): 1485-1489.
Lundrigan, M. D. and R. M. Webb (1992). "Prevalence of ompT among Escherichia coli isolates of human origin." FEMS Microbiol Lett 76(1-2): 51-56.
Meibom, K. L., M. Blokesch, N. A. Dolganov, C. Y. Wu and G. K. Schoolnik (2005). "Chitin induces natural competence in Vibrio cholerae." Science 310(5755): 1824-1827.
Mell JC, Redfield RJ. Natural competence and the evolution of DNA uptake specificity. J Bacteriol. (2014);196(8):1471-83.
Mizusawa, S. and S. Gottesman (1983). "Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein." Proc Natl Acad Sci U S A 80(2): 358-362.
Nishino, T., Morikawa, T., Structure and function of nucleases in DNA repair: shape, grip and blade of the DNA scissors, Oncogene (2002) 21, 9022 – 9032.
Pan, G. and P. D. Sadowski (1992). "Ligation activity of FLP recombinase. The strand ligation activity of a site-specific recombinase using an activated DNA substrate." J Biol Chem 267(18): 12397-12399.
Pollack-Berti, A., M. S. Wollenberg and E. G. Ruby (2010). "Natural transformation of Vibrio fischeri requires tfoX and tfoY." Environ Microbiol 12(8): 2302-2311.
Sadowski, P. D. (1995). "The Flp recombinase of the 2-microns plasmid of Saccharomyces cerevisiae." Prog Nucleic Acid Res Mol Biol 51: 53-91.
Susan T. Lovett, The DNA exonucleases of Escherichia coli, (2003) 4(2).
Savir, Y.,Tlusty, T., RecA-mediated homology search as a nearly optimal signal detection system, Molecular Cell (2010) 40, 388–396.
Santillán M, Mackey MC., Quantitative approaches to the study of bistability in the lac operon of Escherichia coli., J R Soc Interface. (2008) 6;5 Suppl 1:S29-39.
Serebriiskii I.G., Golemis E.A., Uses of lacZ to study gene function: evaluation of β-galactosidase assays employed in the yeast two-hybrid system. Anal. Biochem. 2000;285:1–15.
Studier, F. W. & Moffatt, B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of molecular biology 189, 113–130 (1986).
Sodeinde, O. A., Y. V. Subrahmanyam, K. Stark, T. Quan, Y. Bao and J. D. Goguen (1992). "A surface protease and the invasive character of plague." Science 258(5084): 1004-1007.
Tabor, S. & Richardson, C. C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proceedings of the National Academy of Sciences of the United States of America 82, 1074–1078 (1985).
Villaverde, A. and M. M. Carrio (2003). "Protein aggregation in recombinant bacteria: biological role of inclusion bodies." Biotechnol Lett 25(17): 1385-1395.
Wu R., Zhao M., Li J., Gao H., Kan B., Liang W. Direct regulation of the natural competence regulator gene tfoX by cyclic AMP (cAMP) and cAMP receptor protein (CRP) in Vibrios. Sci Rep. (2015) 7;5:14921.
Weinstock, M. T., E. D. Hesek, C. M. Wilson and D. G. Gibson (2016). "Vibrio natriegens as a fast-growing host for molecular biology." Nat Methods 13(10): 849-851.
Zamenhof, P.J., Villarejo, M. Construction and Properties of E. coli Strains Exhibiting α-Complementation of β-Galactosidase Fragments In Vivo. J Bacteriol. (1972); 110(1): 171–178.
Engler C, Kandzia R, Marillonnet S. 2008. A One Pot, One Step, Precision Cloning Method with High Throughput Capability. PLOS ONE 3: e3647.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0003647 (October 18, 2018)
Iverson SV, Haddock TL, Beal J, Densmore DM. 2016. CIDAR MoClo: Improved MoClo Assembly Standard and New E. coli Part Library Enable Rapid Combinatorial Design for Synthetic and Traditional Biology. ACS Synthetic Biology 5: 99–103.
https://doi.org/10.1021/acssynbio.5b00124 (October 18, 2018)
Lee ME, DeLoache WC, Cervantes B, Dueber JE. 2015. A Highly Characterized Yeast Toolkit for Modular, Multipart Assembly. ACS Synthetic Biology 4: 975–986.
https://doi.org/10.1021/sb500366v (October 18, 2018)
Moore SJ, Lai H-E, Kelwick RJR, Chee SM, Bell DJ, Polizzi KM, Freemont PS. 2016. EcoFlex: A Multifunctional MoClo Kit for E. coli Synthetic Biology. ACS Synthetic Biology 5: 1059–1069.
https://doi.org/10.1021/acssynbio.6b00031 (October 18, 2018)
Patron NJ, Orzaez D, Marillonnet S, Warzecha H, Matthewman C, Youles M, Raitskin O, Leveau A, Farré G, Rogers C, et al. 2015. Standards for plant synthetic biology: a common syntax for exchange of DNA parts. New Phytologist 208: 13–19.
https://nph.onlinelibrary.wiley.com/doi/abs/10.1111/nph.13532 (October 18, 2018)
Pollak B, Cerda A, Delmans M, Álamos S, Moyano T, West A, Gutiérrez RA, Patron N, Federici F, Haseloff J. 2018. Loop Assembly: a simple and open system for recursive fabrication of DNA circuits. bioRxiv: 247593.
https://www.biorxiv.org/content/early/2018/01/18/247593 (October 18, 2018)
Sarrion-Perdigones A, Falconi EE, Zandalinas SI, Juárez P, Fernández-del-Carmen A, Granell A, Orzaez D. 2011. GoldenBraid: An Iterative Cloning System for Standardized Assembly of Reusable Genetic Modules. PLOS ONE 6: e21622.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0021622 (October 18, 2018)
Sarrion-Perdigones A, Vazquez-Vilar M, Palací J, Castelijns B, Forment J, Ziarsolo P, Blanca J, Granell A, Orzaez D. 2013. GoldenBraid 2.0: A Comprehensive DNA Assembly Framework for Plant Synthetic Biology. Plant Physiology 162: 1618–1631.
http://www.plantphysiol.org/content/162/3/1618 (October 18, 2018)
Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S. 2011. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLOS ONE 6: e16765.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0016765 (October 18, 2018)