Team:Queens Canada/Demonstrate
Results
Experiments: This summer QGEM accomplished a lot of work in the laboratory! We have 1) validated the Engineered NanoLuc luciferase as an choice reporter in our intein-based biosensor construct, 2) We characterized the insolubility of a small-molecule triggered intein splicing system in E. Coli, 3) We validated the nuclear receptor ligand binding as a sensitive domain for analyte detection through the successful assembly of a reagent-less, and continuous FRET-based biosensor with the ability to discern between agonist and antagonists.
1) Validation of NanoLuc Luciferase for application in our Diagnostic Pacifier
To create our desired portable intein-splicing biosensor, we needed to select a signal that could be measured and correlated
to the amount of analyte present. Additionally, we had the challenge of selecting a reporter that could be produced and quantified
very portably. Our first idea was to use a fluorescent reporter, however with fluorescence, a fluorophore must first be excited by
a light, and then a light is given off that needs to filter and its intensity measured. Fluorimeter are bulky devices, and after
further investigation, it become apparent that it would be possible the machinery down to fit with a pacifier. However, during our
investigations we learned about the elements needed to build a luminometer and found that it would be entirely feasible for us to
produce a simplified photon-counter device that could fit into a pacifier! (See Hardware section).
To best improve our chances of being able to produce a signal which our portable luminometer could detect we investigated various
the relative strength of luciferases. We ultimately decided on testing the novel and very bright NanoLuc Luciferase as the quantitative
reporter for our construct. NanoLuc® developed by Promega is a luciferase derived via directed evolution from the luminous shrimp,
Oplophorus gracilirostris [1]. The enzyme was obtained from deep-sea shrimp and optimized following the discovery of a novel substrate,
furimazine, which allows for the production of visible light with less background activity than other luciferases [1,2]. NanoLuc® is a
relatively small, a19.1 kDa monomeric protein that is both soluble and ATP-independent [1]. Compared to firefly (Lampyridae) and sea
pansy (Renilla) luciferases, this novel protein offers many advantages reflected by its increased stability, smaller size, and >150-fold
increase in luminescence [2]. The unique characteristics of this enzyme construct combined with its high luminescence activity allow for
the production of a very sensitive diagnostic assay.
By generous donation from Promega, we received the promoter-less vector pNL1.1. We then cloned an Anderson Constitutive promoter
(Part:BBa_J23100) into the MCS of pNL1.1. To test our construct, we grew an overnight liquid culture of E. Coli expressing the plasmid
construct. We pelleted 2-3ml of liquid culture, resuspended in a lytic buffer with lysozyme and protease inhibitor for 30 mins, before
sonicating for 10 seconds the placing on ice, twice. We then pelleted the insoluble fraction, collected the supernatant and performed a
Luciferase Assay, using Nano-Glo® Luciferase Assay System. Relative light units were measured in a Lumistar Galaxy plate reader. We
then took an initial measurement of Relative Light Unit and found a strong result of 38598.2±3385.40 RLU (n=4), compared to 103.5±33.6
RLU (n=5), with our negative control at peak luminescence. Since the ultimate goal of using NanoLuc will be to produce a signal for
diagnostic purposes, we sought to further characterize the time-course of NanoLuc Luciferase after the addition of substrate. Through
taking measurements every 0.05 seconds, we were able to produce a curve demonstrating the time till plateau of NanoLuc after addition
of substrate. Within 10 seconds, NanoLuc had reached its plateau, therefore validating NanoLuc would be an excellent reporter protein
for application in our intein-based diagnostic biosensor.
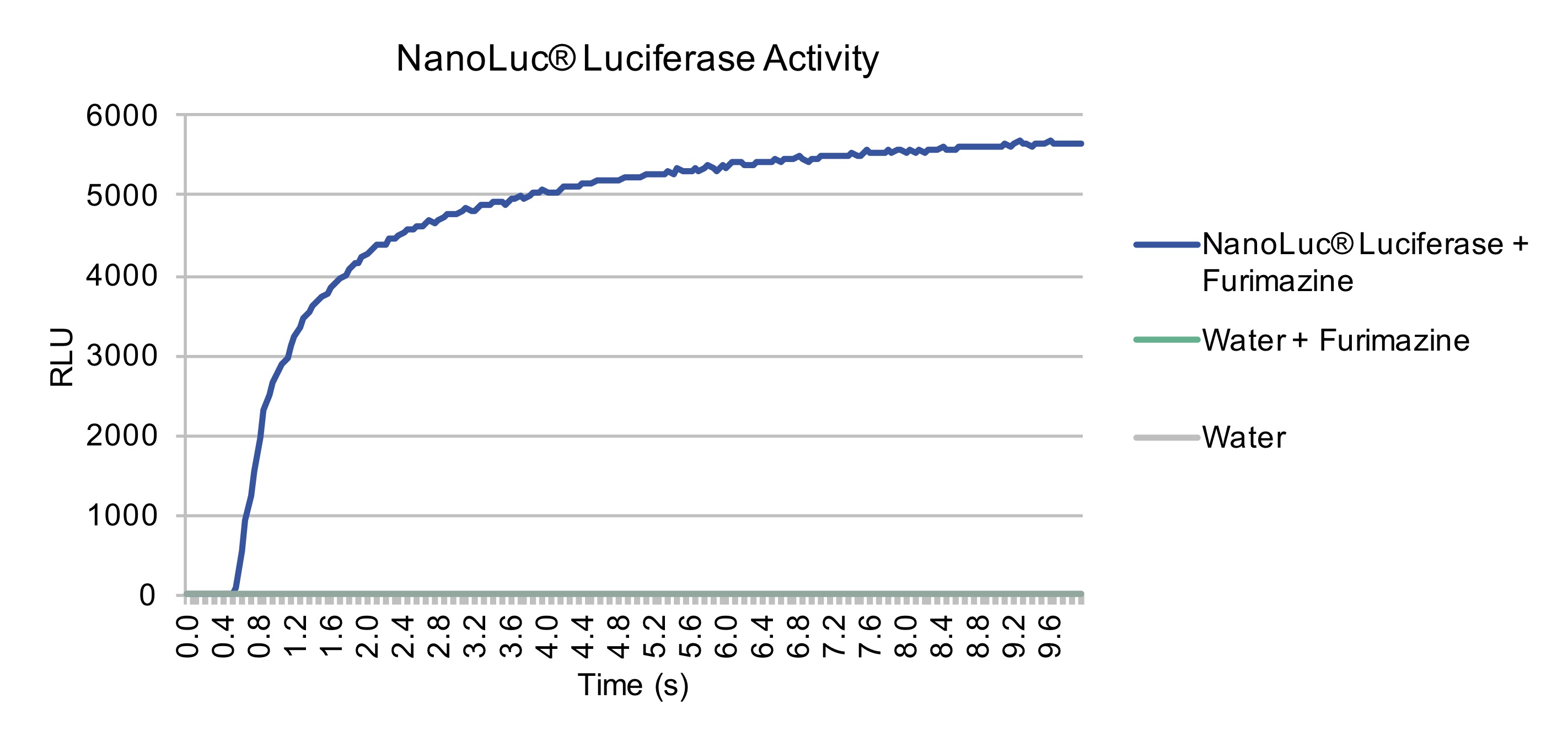
We next sough to characterize the sensitivity of our portable luminometer to the luminescence produced by NanoLuc. The portable luminometer measures differently than our labs luminometer, as the Lumistar Glaxay luminometer in our lab takes an instantaneous measurement of light, in relative light units. Whereas, our built luminometer measures photons/second. Therefore, the two devices cannot be directly compared. For calibration of our luminometer we first confirmed the sample to be luminescing by a measurement with the Lumistar Galaxy Luminometer, we then measured samples diluted 1, 2, and 4-fold and found our luminometer was capable of measuring a linear relationship between the concentrations of our sample. Thus confirming our luciferase construct was functional and rised to plateau quickly, our built portable luminometer was accurate across a range of concentrations, and that Nanoluc Luciferase could serve as a viable signal for our diagnostic assays.

2) Characterization of insolubility of a small-molecule triggered intein splicing system in E. Coli
The goal of this year’s project was to create a biosensor that would luminesce in the presence of cortisol, giving our team the ability to quantify the hormone in saliva. The idea was ignited from an small-molecule triggered intein previously developed by Buskirk et al (2004). In this article, the authors inserted the ligand binding domain of the Estrogen Receptor into the RecA intein, disrupting its ability to perform auto-splicing, as found in nature. Splicing was only restored after the addition of the estrogen antagonist 4-hydroxytamoxifen (4-HT), and rounds of mutagenesis. The construct was then inserted into variety of split extein contexts, such as kanamycin resistance, and GFP, and found to splice together the split proteins halves and produce the mature protein, in a dose-responsive manner to 4-HT addition.
We sought to expand on the detection capabilities of this system, by creating a modular system, where both the ligand binding domain, and the extein could be replaced. The ultimate goal of the project was to create a biological sensor for the detection of cortisol. We would achieve this by swapping the estrogen receptor ligand binding domain for the glucocorticoid receptor, and replace the kanamycin resistance extein with the bright luciferase, NanoLuc.
Nuclear receptors such as estrogen and glucocorticoid share large homology in their DNA sequence, and 3D strucutre. The generic LBD of nuclear receptors is comprised of 12 a-helices and 4 small β-sheets that fold into a three-layer helical domain. Upon binding with an antagonist or agonist, the receptors undergo a conformational change. The conformational change is highlighted in the movement of helix 12. Our team modeled a variety of linkers for the glucocorticoid, which would connect the halves of the RecA intein upon cortisol binding We wanted linkers long enough to be able to wrap around the molecule upon conformational change and binding with the correct ligand, however the linkers needed to be rigid and short enough, not to induce auto-splicing without the need of ligand binding. The following linkers were designed based on the optimal angstrom distances and flexible design. Our dry lab team calculated that for a rigid linker, the shortest possible distance needed when the conformational change was initiated was 8 angstrom, and 20 angstrom for a more flexible linkers. We tried out three different linkers for our glucocorticoid construct, as we were unsure how rigid or flexible the linker needed to be to ensure splicing. Once the linkers were chosen, we performed molecular modelling to ensure that the two inteins halves would come into close enough contact to initiate splicing, and thus produce luminescence. The molecular dynamic simulations verified that the linkers: N-terminal: GPGSGS, C-terminal: GASGSG, were in fact long enough for the intein to come in contact, and they even formed a favourable interaction in just 10 nanosecond of simulation time. (see Molecular Dynamic Simulations her). Armed with knowledge gained in our dry lab, we set out to recreate the results by Buskirk et al. (2004), and develop new constructs based on the glucocorticoid receptor.
For proof of concept, we conducted experiments on these constructs in Eschiceria coli. Although the original paper utilized yeast, we set out to test the ability of the 4-HT dependent intein to splice in bacteria, in efforts to improve its characterization, and due to the relative ease of working with novel genetic constructs in yeast. We firstly constructed the Kanamycin-4HT-Intein, and Kanamycin-GR-Intein in the expression vector pET16b via gibson assembely with overlapping sequence, and then transformed DH5-alpha. We picked colonies, grew liquid cultures, mini-prepped, and performed diagnostic digests to confirm our insert was present.


We then transformed our construct into BL-21 DE3, a strain of E. coli that is important for protein expression, and induced expression with IPTG. To test intein splicing, we made agar plates containing either 1) Ampicillin, as a positive control, 2) Kanamycin, as a negative control 3) Kanamycin and 4-HT. We plated the BL21 DE3 and found that no bacterial colonies grew on the Kanamycin alone, or Kanamycin and 4-HT containing plates, but there was growth on the Ampicillin containing plates. If the iintein was in fact splicing caused by 4-HT, we would expect to see growth on the Kanamycin and 4-HT containing plate. Therefore, this demonstrated that the intein construct was not splicing and Kanamycin Resistance was not being produced.

To discern why the intein was not splicing we ran a SDS-page on induced BL21-DE3 expression. We grew 2 liquid cultures of BL21 DE3 containing the intein, induced expression of T7 RNA polymerase with IPTG, then incubated both for 4 hours at 37C. We then added 10μm 4-HT to one of the liquid cultures and incubated at 37C for an additional 1 hour to allow splicing of the intein to occur.
The team lysed the cells by addition of lysozyme and protease inhibitor containing lysis buffer, followed by sonication for 10 seconds. 10 seconds would provide sufficient enough time for splicing without destroying the cells, or having the liquid bacteria overheat. We then collected 1ml of the mixed fraction and stored at -20C. The remainder of the fraction were centrifuged at 8k RPM for 5 minutes and collected 1ml of the soluble fraction. The remainder of the soluble fraction was discarded, and the insoluble fraction was resuspended, and 1 ml was collected as the insoluble fraction.
We then ran a SDS-page to determine protein expression, and if splicing had occurred.

The SDS page informed us that the majority of our protein product was found in the pellet and not the supernatant. This indicated that our construct was not soluble, and would not be able to splice.
We subsequently found one paper that had previously utilized dose-dependent intein splicing in bacteria, however this paper further tagged intein constructs with a maltose-binding protein.[3] Therefore, this further demonstrates the insolubility of small-molecule triggered intein systems, such as the one we sought to devleop.
Our next step was to try and increase our protein’s solubility. Through a collaboration with the Stonybrook iGEM team, we decided to try directed evolution through error-prone PCR. We created primers for the insert and the backbone, and added MnCl¬2 to our PCR reaction to induce random mutagenesis. We then tried Gibson assembly and transformation of the mutagenized product to see if any insert or backbone had worked. If colonies grew on kanamycin and cortisol, we know that splicing worked. The colonies would then be sampled and plated on kanamycin, if they grew, then the intein was not selective for just ligand binding. Unfortunately, we saw no evidence of the error prone PCR being successful after numerous attempts.
In the future, our constructs solubility could be increased through error-prone PCR, with a careful selection process. As well, we discussed the possibility of adding a maltose binding domain to our protein and increasing its solubility. In E. coli, maltose binding protein, can be used to promote solubility of the protein that it is fused with.
Since the intein construct previously worked in yeast, we also believe our construct could be successfully constructed and spliced upon ligand binding in this organism.
3) Validation of the nuclear receptor ligand binding as a ligand binding domain producing pronounced conformational changes through the successful assembly of a reagent-less, and continuous FRET-based biosensor with the ability to discern between agonist and antagonists.
We took advantage of this conformational change upon ligand binding to develop a glucocoticoid biosensor capable of detecting agonists, or antagonists of the receptor utilizing fluorescent resonance energy transfer (FRET). FRET is a process in which energy is transferred from an excited donor fluorescent reagent to an acceptor fluorescent reagent. FRET requires that the emission spectrum of the donor protein and the absorbance spectrum of the acceptor protein overlap adequately, so that light emitted from the donor can excite the acceptor. FRET is highly sensitive to the distance between the first and second fluorescent reagents. For example, FRET varies inversely with the sixth power of the d stance between the first and second fluorescent reagents.
Nuclear receptors share a largely homologous structure in 3D, where binding of a ligand which is an agonist changes the receptor from an inactive state of the receptor into an activate one, capable of inducing transcription. This changes is mediated through conformational changes in the proteins quaternary structure. The terminal helix 12 portion of the LBD undergoes pronounced movement to bring about this effect. The binding of antagonist ligands to nuclear receptors produces a different conformational change that prevents induction of transcription. In the absence of ligand, N- and C- terminals of the ligand binding domain are in a proximity such that energy transfer from a fluorescent protein donor to a fluorescent protein acceptor can occur. If agonist is present, a conformational change occurs impacting the distant dependent energy transfer between the fluorescent proteins, which can be detected by a change in FRET. Antagonists can also be identified by virtue of their ability to create a remarkably different change in conformation, resulting a in FRET efficiency distinct of the unbound, and antagonist bound conformations.

We connected the FRET donor, acGFP1 to the N-terminal of the GR LBD, separated by a short 4AA linker, and we connected the FRET acceptor, mCherry to the C-terminal of the GR LBD, separated by a short 11AA linker. The sequences of acGFP1, the GR LBD, and mCherry were all ordered in 3 separate parts from Integrated DNA Technologies as double stranded gblocks with >15 basepair overlaps, and N- and C-terminal overlaps for the vector pET-16b. pET-16b was plasmid miniprepped from DH5-a E. coli, and then digested with XhoI and BamHI restriction endonucleases to produce a linearized fragment. The linearized pET-16b, acGFP1, the GR LBD, and mCherry gblock DNA fragments were then mixed at equal molar ratios, and then joined into a circular DNA plasmid by Gibson Assembly. We confirmed the presence of the insert using a diagnostic digest with NdeI.

To determine if FRET was occuring between the fluorescent proteins, we incubated BL21 DE3 expressing the FRET construct in pET-16b, with various concentrations of IPTG. After 4 hours of incubation at 37C, we lysed cells by addition of lysozyme and protease inhibitor containing lysis buffer, followed by sonication for 10 seconds. We then centrifuged the solution, and collected the soluble protein containing supernatant. We put samples of the soluble cell lysate into a SpectraMax iD5 Multi-Mode Microplate Reader set for excitation at 475nm, the maximal excitation of acGFP1, and the emission filter to 610nm, the emission wavelength of mCherry. We observed a large amount of FRET was occurring, as seen by the emission at 610nm.

We next sought to test if a change in conformation occurred in response to ligand binding. Colonies were then picked and grown for 4 hours in LB broth containing ampicillin and 0.1M IPTG shaking at 250rpm at 37C. 1ml of liquid culture was then aliquoted into 3 separate tubes, to the first was added 100uM cortisol dissolved in 10ul of ethanol, to the second was added 100uM miferpistone dissolved in 10ul of ethanol, to the third was added 10ul of 100% ethanol. The 1ml liquid cultures containing cortisol, mifepristone, and ethanol were then incubated at room temperature for 30 minutes. The 1ml liquid cultures were then pelleted by centrifugation for 5 minutes at 8000 rpm. The supernatant was tossed, and the pellet was then smeared on individual Microscope slides. The microscope slides were then viewed with a Leica SP2 Laser Scanning Confocal Microscope with a laser line set to 488nm. Fluorescence detection was observed at 505nm, and 610nm. Numerous high quality images were taken of each slide at both fluorescence detection wavelengths for multiple regions of interest. Fluorescence was then quantified using imageJ selecting the same 4 regions of interest per image and measuring average intensity in each Region of Interest. Average FRET efficiency in a given condition was then calculated using the formula: Efficiency = (Fluorescence Acceptor / (Fluorescence Acceptor + Fluorescence Donor)).



Therefore, we have produced a biosensor capable of detecting the hormone cortisol, and producing a signal distinct of the unbound conformation. Additionally, our construct was capable of detecting agonists and antagonists of the GR receptor, and produce pronounced differences in FRET efficiency, which would be used to discern an agonist vs an antagonist bound. Given the therapeutic importance of steroid hormones such as estrogen, cortisol, progesterone, synthetic agonists (e.g dexamethasone, and prednisone), and antagonists of nuclear receptors (e.g tamoxifene, and miferpristone), this provides a potential method to identify pharmaceutical compounds affecting nuclear receptors.

In the absence of agonist, the first fluorescent reagent, bound to the nuclear receptor or ligand binding domain thereof, will start in close proximity to the second fluorescent reagent such that FRET will be observed. In the presence of agonist, conformational changes occurs thus bringing the first and the second fluorescent reagents further apart, allowing FRET to occur less and the change in FRET can be observed. In the presence of antagonist, conformational changes occurs thus bringing the first and the second fluorescent reagents even further apart than the agonist bound conformation, allowing FRET to occur even less and the change in FRET can be observed.
References
- C. G. England, E. B. Ehlerding, and W. Cai, “NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence., Bioconjug. Chem., vol. 27, no. 5, pp. 1175–1187, 2016."
- . Boute, P. Lowe, S. Berger, M. Malissard, A. Robert, and M. Tesar, “NanoLuc Luciferase - A Multifunctional Tool for High Throughput Antibody Screening., Front. Pharmacol., vol. 7, p. 27, 2016.
- L. G. G. C. Verhoef, M. Mattioli, F. Ricci, Y.-C. Li, and M. Wade, “Multiplex detection of protein–protein interactions using a next generation luciferase reporter,” Biochim. Biophys. Acta - Mol. Cell Res., vol. 1863, no. 2, pp. 284–292, Feb. 2016.
- A. R. Buskirk, Y.-C. Ong, Z. J. Gartner, and D. R. Liu," “Directed evolution of ligand dependence: small-molecule-activated protein splicing., Proc. Natl. Acad. Sci. U. S. A., vol. 101, no. 29, pp. 10505–10, Jul. 2004.