Team:UBonn HBRS/Description/SELEX
SELEX
SELEX (Systemic Evolution of Ligands via EXponential Selection) is the process of identifying specific aptamers from a random pool of DNA or RNA that have a high affinity to a specific target molecule. The process of SELEX can be subdivided into three stages.
Contents
Finding the right pool of nucleic acids
The first stage is the selection of the right pool of single-stranded nucleic acids to start with. For the further application of the aptamer, you need to decide whether you want it to be composed of ssRNA or ssDNA. One weak spot of aptamers is their susceptibility to nucleases. Nucleases are restriction enzymes that digest oligonucleotides and they are omnipresent. Especially, RNA is very instable in most environments. To circumvent this instability, it is possible to edit RNA as well as DNA to make them resistant to any nucleases. Modified nucleotides such as 2’-amino pyrimidines, 2’-fluoro pyrimidines or 2’-O-methyl ribose purines can be incorporated into the oligonucleotides to render them resistant against nucleases.¹ However, as we want our end product to be as cheap as possible, we chose an unmodified DNA pool. In general, DNA is more stable than RNA, so we hope that our aptamers will not be degraded by salivary nucleases. Otherwise, we have to modify them afterwards. Another point is the length of the oligonucleotides in the DNA library. Again, we plan in the cost factor and thus want a rather short sequence to start with. We decided on a library of 80-mers, called D3 library provided by Ella Biotechnology, Martinsried. These oligonucleotides are build up from 18nt as a forward primer, followed by a sequence of 43 random nucleotides and ending with a 19nt sequence for the reverse primer to bind to. The library is designed to have up to 10¹⁵ different nucleotide sequences.
SELEX
In order to select only those aptamers that specifically bind to our target, it is necessary to establish a method to separate binding from non-binding oligonucleotides. Together with the boom of aptamers, many new techniques for SELEX have been established. It is possible to bind your target to affinity beads or nitrocellulose membranes and wash off non-binders or capillary electrophoresis may be used to select for aptamers, only to name a few.² We decided on using magnetic affinity beads to fix our target proteins. As we tagged our proteins with a 6xHis-tag, we can bind them to magnetic cobalt beads to fix them with a magnet when needed. The whole process of our SELEX is illustrated in Figure 1. At first, we bind our protein of interest to the beads. In a next step, we incubate the beads with the ssDNA library at 37°C in a saliva-like buffer to be as close to the conditions in the mouth as possible. After the incubation, the beads are fixed to the wall of the reaction tube by applying a magnetic field to be able to wash off unbound ssDNA. In a next step, the proteins together with all bound aptamers are eluted from the beads by adding an imidazole-solution. The aptamers bound to the protein are then amplified in a PCR reaction. After the PCR reactions, the double-stranded oligonucleotides need to be rendered in a single-stranded form again. To this end, one of the primers is phosphorylated at its 5’-end so it may serve as a target for a lambda exonuclease that digests that DNA strand leaving only ssDNAs again. This ssDNA now serves as a pool for the next round.
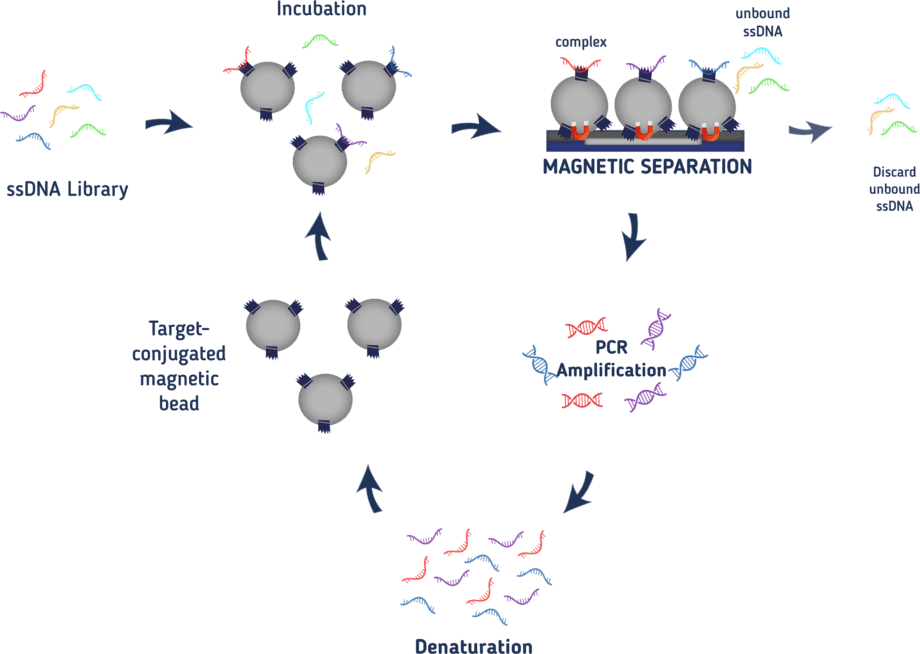
With every round, more and more oligonucleotides with low binding-affinity for our protein of interest will be discarded leaving only strong binding aptamers at the end. From the third round on, the amount of protein used in the SELEX is decreased with every round to increase the pressure for low-affinity binding proteins. Also from the third round on, the ssDNA will be incubated with the magnetic cobalt beads without any protein bound to it before the actual incubation with the protein. This way, aptamers binding to the magnetic beads and not the protein of interest are eliminated. To also eliminate aptamers that unspecifically bind to random proteins, bovine serum albumin is added to the buffer to wash off those aptamers. The buffer also contains salmon sperm to increase the selection pressure as this DNA may also interact with the proteins instead of our aptamers. Only aptamers with a high binding affinity will be able to compete. After 8-10 rounds a specific aptamer should be detected.
Characterization of the aptamer
As a final step, the oligonucleotides from every round will be analyzed by next-generation sequencing to track the separation process of non-binding and binding aptamers and to find out the exact sequence of our final aptamer. Moreover, the effect of the aptamer on the function of our proteins will need to be tested as described in the chapter ‘Enzyme assay’. Finally, the exact binding affinity of the aptamer will need to be determined.
Experimental approach
Establishing a new SELEX experiment with a new target always needs some preceding tests. As we were not able to express our proteins of interest yet, we ordered a fragment of GTFC to use it for the establishment of our enzyme assays and to perform SELEX on it. This protein contains the catalytic domain from the amino-acid residues 244–1163.The protein has a molecular weight of 98.9 kDA.³’⁴
Saliva-like binding buffer
One of the first things to consider upon SELEX is which buffer to use during the binding of the aptamers to your protein. As we want to our condition as close as possible to the mouth, we chose a saliva-like buffer. We decided to use a modification of the Mondelli buffer from the paper ‘Electrochemical behavior and pH stability of artificial salivas for corrosion tests’.⁵ It has, compared to other saliva-like buffers in the paper, the best pH stability. It originally has a pH of 5.3 which is lower than in physiological saliva. So, we decided to modify it and adjusted the pH to 6.3 by lowering the amount of the acid NaH₂PO₄. This is a compromise between the pH-optimum of the enzyme (which is 6.0, according to the manufacturer) and the pH of actual human saliva (between 6.2 and 7.66).⁶ To avoid precipitation of CaHPO₄, the calciumchloride contained in the buffer isn’t added at once with the other substances. We prepare a 10x stock solution (6mg/ml, in ddH₂O) which is added directy before use to the rest of the buffer. The final modified Mondelli buffer has the following concentrations with a pH of 6.4:
| Substance | Mass concentration [mg/l] | Molar concentration [mM] |
|---|---|---|
| NaCl | 500 | 8.56 |
| KCl | 500 | 6.71 |
| CaCl₂ | 600 | 5.41 |
| Citric acid · H₂O | 5.5 | 0.026 |
| NaH₂PO₄ · H₂O | 346 | 5.65 |
| Urea | 1000 | 16.7 |
| (NH₄)₂SO₄ | 300 | 2.27 |
| NaHCO₃ | 100 | 1.19 |
For the SELEX experiments, this buffer will be supplemented with 0.1mg/ml BSA, 0.1mg/ml Salmon sperm and 0.1% Tween.
Inhibiton of D3 amplification PCR by GTFC
First, the influence of the protein or the saliva-like-binding-buffer (Mondelli-buffer) on the PCR amplification of the oligonucleotides needs to be evaluated. If it would inhibit the PCR, protein and ssDNA would need to be separated before the amplification by methods like phenol-chlorophorm extraction. Though the saliva-like-buffer inhibits the PCR amplification of the D3 library (Figure 2), the protein alone with imidazole and ddH₂O did not inhibit the PCR. (Figure 3) Upon addition of the protein, the D3 library amplification only needs two more cycles to amplify the same amount of DNA. We can thus elute the protein with bound ssDNA from the beads with ddH2O and imidazole and use the eluate for the PCR amplification.
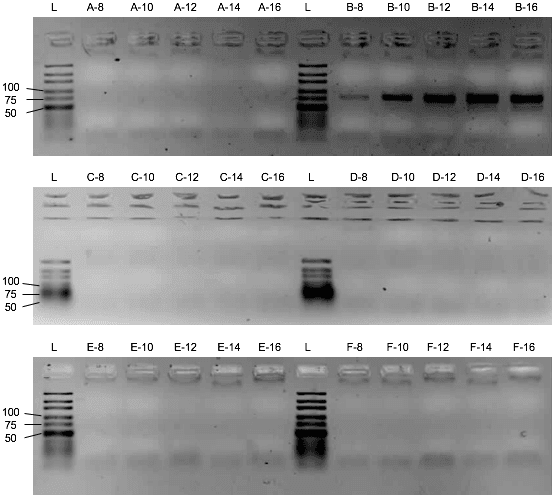
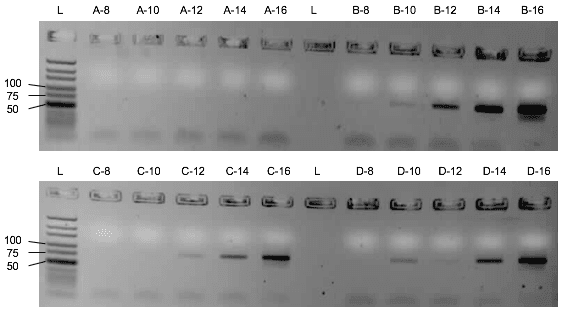
Assessment of the binding capacity of the cobalt beads
In a second step, we controlled the binding capacity of the cobalt beads for our protein. After coupling different amounts of GTFC to the magnetic cobalt beads it turned out that 1 mg of beads is able to bind approximately 40 µg of GTFC as indicated in the provider’s instructions. When we added 67.2 µg of GTFC to 1 µg of beads, some of the protein was not bound to the beads anymore, but showed up in the first flow-through. For 13.4ng of GTFC, a weaker protein band showed up upon elution, so at this point the beads were not saturated yet. This means that the optimal binding capacity is between 13.4µg and 67.2µg per µg beads. (Figure 4)
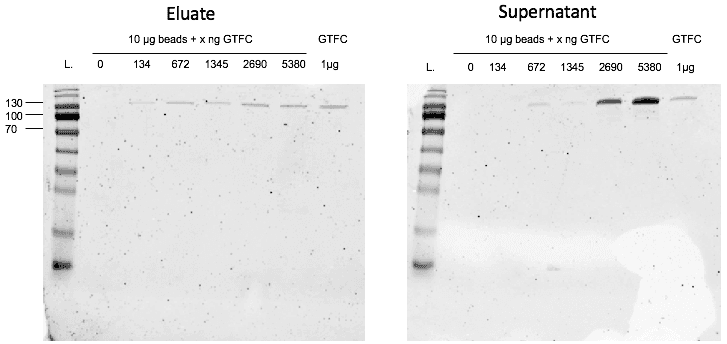
Unfortunately, we ran out of time to perform the first SELEX before the end of this year’s project.
Protocols used
Changing the buffer of the commercially obtained GTFC
As the GTFC was delivered in a glycerol-supplemented stock containing 200 mM imidazol, we first had to exchange the buffer to be able to couple the protein to the magnetic cobalt beads.
For this we used the Amicon Ultra-0.5 Centrifugal Filter Unit with Ultracel-10 membrane (Merck, UFC501096) according to the manufacturer’s manual using the Mondelli buffer described in ‘Enzyme assay’.
D3 library amplification
| Reagent | Final concentration |
|---|---|
| 5x GoTaq buffer | 1x |
| MgCl₂ (25 mM) | 2 mM |
| dNTPs (25 mM each) | 200 µM |
| Fwd. Primer (100 µM) | 0.5 µM |
| Rev. Primer (100 µM) | 0.5 µM |
| GoTaq Polymerase (5 U/µl) | 1 U |
| DNA template (1 fmol) | 1 fmol/100 µl |
| ddH₂O | Ad 100 µl |
Forward primer: 5'-GCT GTG TGA CTC CTG CAA-'3
Reverse primer: 5'-Phos-GGA GAC AAG ATA CAG CTG C-3'
Master mixes of 100µl were always prepared that were separated into 5 reaction tubes à 20µl to take out on tube after every indicated cycle. For the first inhibition test, following mastermixes were prepared: No template control=no DNA template; 1 fmol DNA template and ddH₂O; 1 fmol DNA template and Mondelli buffer instead of ddH₂O; 1fmol DNA template and 1:1 Mondelli buffer:ddH₂O; 1 fmol DNA template and Mondelli buffer instead of ddH2O plus 3 µg GTFC; 1fmol DNA template and 1:1 Mondelli buffer:ddH2O plus 1.5µl GTFC. For the second inhibition test, following Mastermixes were prepared: No template control=no DNA template; 1 fmol DNA template and ddH₂O; 1 fmol DNA template and ddH₂O plus 13,5µg GTFC (diluted in ddH₂O (+300 mM imidazole)); 1 fmol DNA template and ddH2O plus 8,2µg GTFC (diluted in ddH₂O (+300 mM imidazole)).
PCR program: 1) 5’ at 95°C 2) 1’ at 95°C 3) 1’ at 64°C 4) 1’30’’ at 72°C 5) 3’ at 72°C 5) ∞ at 10°C; for every cycle step 2)-4) were repeated.
Binding assay
GTFC was coupled to magnetic cobalt beads (Dynabeads™ His-Tag Isolation and Pulldown, Thermo Fisher Scientific, 10103D) according to the manufacturer’s manual. However, as a binding buffer we used our Saliva-like-binding-buffer (Mondelli buffer). As an elution buffer, we used a 300mM imidazole solution in ddH₂O.
SDS-PAGE
For the separation of GTFC, we used a 10% SDS-PAGE. The proteins were separated at The proteins on the gel were then stained in Coomassie Blue (30% acidic acid, 10% methanol, 1 g/l coomassie G-250) for 10 min, briefly boiled in the Coomassie Blue for a quicker staining and cooled off. The gel was destained with a 15% acidic acid destaining solution by briefly boiling it up and changing the solution three times every ten minutes. The gels were then destained under slight shaking overnight.
References
- 1. Nimjee, S. M., White, R. R., Becker, R. C. & Sullenger, B. A. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 57, 61–79 (2017).
- 2. Gopinath, S. C. B. Methods developed for SELEX. Anal. Bioanal. Chem. 387, 171–182 (2007).
- 3. Ito, K. et al. Crystal Structure of Glucansucrase from the Dental Caries Pathogen Streptococcus mutans. J. Mol. Biol. 408, 177–186 (2011).
- 4. Ito, K. et al. Crystallization and preliminary X-ray analysis of a glucansucrase from the dental caries pathogen Streptococcus mutans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1086–1088 (2010).
- 5. Queiroz, G. M. O. De, Silva, L. F., Ferreira, Jo. T. L., Gomes, J. A. D. C. P. & Sathler, L. Electrochemical behavior and pH stability of artificial salivas for corrosion tests. Braz. Oral Res. 21, 209–215 (2007).
- 6. Baliga, S., Muglikar, S. & Kale, R. Salivary pH: A diagnostic biomarker. J. Indian Soc. Periodontol. 17, 461–465 (2013).