Team:USMA-West Point/Results
Project
Results
Construction of hOR1D2 expression plasmid
A mammalian expression plasmid containing hOR1D2 was previously obtained by the laboratory for other eNOSE-related projects. Since this project requires hOR1D2 expression in bacteria, however, the hOR1D2 coding sequence needed to be shuttled to a bacterial expression plasmid, the pET-21A expression vector. To amplify the hOR1D2 coding with restriction enzyme sites compatible for the pET-21A vector, the following oligonucleotides were ordered (from ThermoFisher):
Forward primer: 5’-ATCGATCG GAATTC ATGGATGGAGGCAACCAGAGT-3’
Reverse primer: 5’- ATCGATCG AAGCTT TCATGTCAGCCTCTTAAAGTGTTTATCT-3’
The primers were designed to place an EcoRI restriction enzyme site on the 5’ end of the coding sequence (EcoRI site is underlined in the forward primer sequence; ATG translation initiation codon is in bold). The primers were also designed to introduce a HindIII restriction enzyme site on the 3’ end of the coding sequence (HindIII site is underlined in the reverse primer sequence; translation Stop codon is in bold).
Together, these primers were designed to directionally orient the hOR1D2 coding sequence in frame with the open reading frame established in the pET-21A plasmid (see Figure 1).
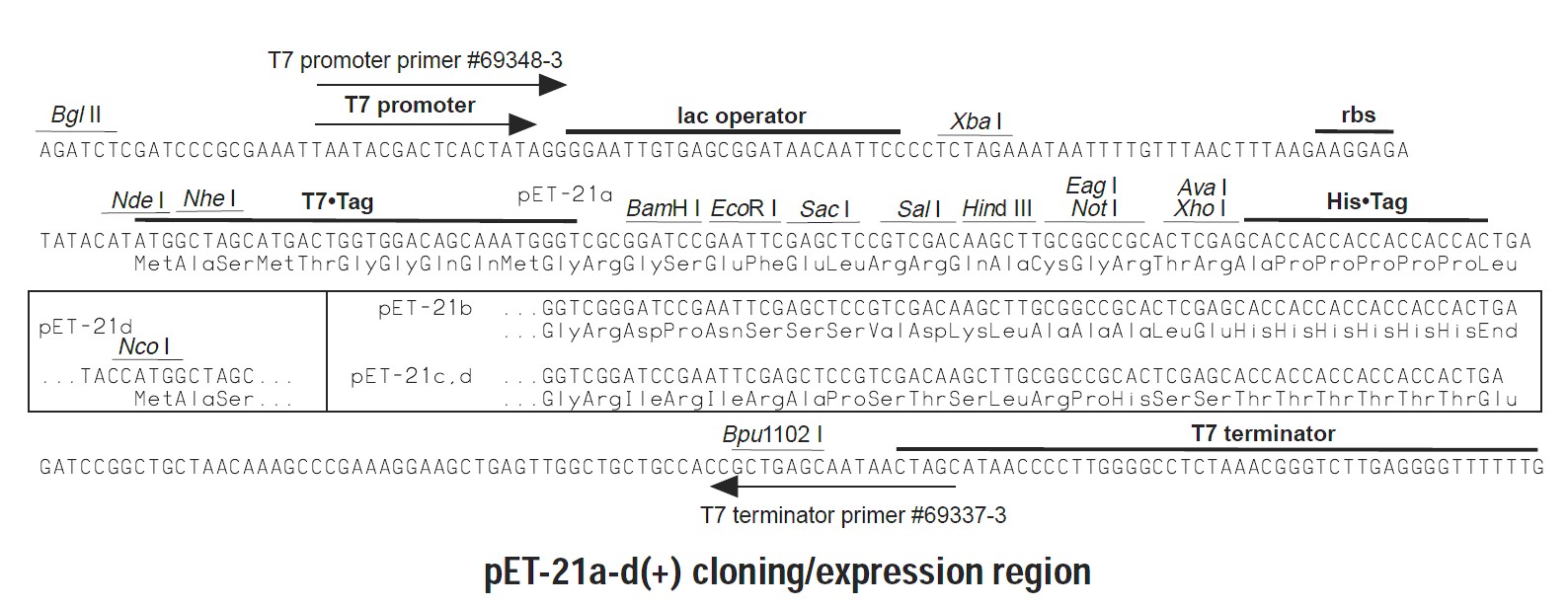
Figure 1: Multiple cloning site and open reading frame for pET-21A plasmid (figure source Nova Lifetech Inc).
Following the PCR reaction to amplify hOR1D2 coding sequence with the primers shown above, an agarose gel was run to confirm amplification of the coding sequence (Figure 2). This gel shows a single band at just under 1kb, which is consistent with the predicted 967bp length based on sequence information.
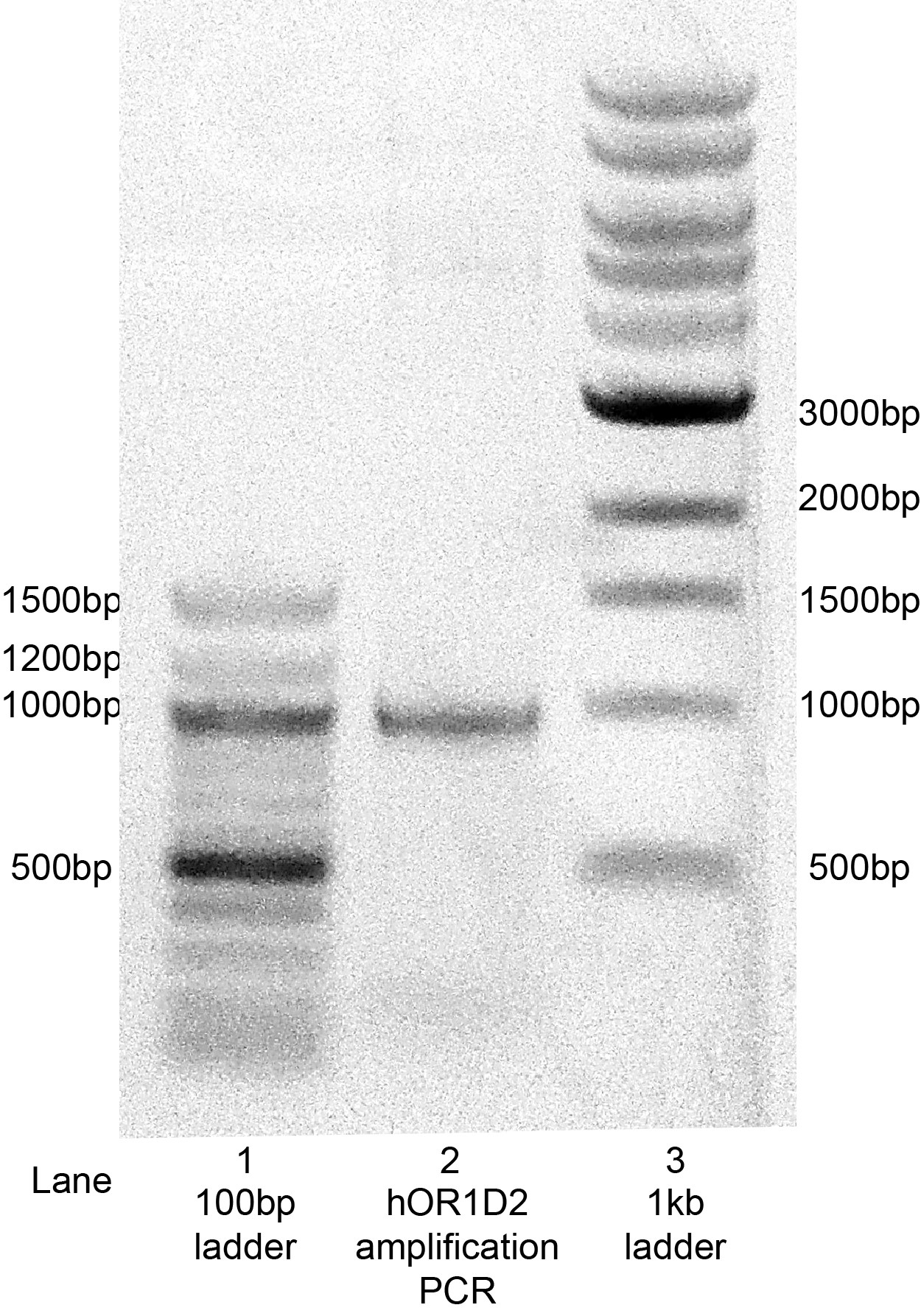
Figure 2: Amplification of the hOR1D2 coding sequence by PCR
This PCR product was purified, and then digested with EcoRI and HindIII overnight. The following morning, the plasmid was purified from digestion reaction, and eluted in a 30uL volume of water. The NanoDrop measured that the purified, digested hOR1D2 insert was at 55ng/uL.
A 1uL aliquot of 250ng/uL stock of pET-21A was used to transform DH5alpha E.coli. A single colony from this transformation was used to start an overnight 5mL culture. A half milliliter of this overnight culture was used to make glycerol stock that was stored in the -80C freezer, and the remaining portion of the culture was used to produce a 50uL stock of pET-21A in water at 400ng/uL.
To prepare the pET-21A, 5ug was digested with EcoRI and HindIII overnight. The following morning, 1uL of Antarctic Alkaline Phosphatase was added to the digestion reaction (to dephosphorylate free 5’ phosphate groups on the digested plasmid and reduce background colonies), and incubated for an additional hour, after which the reaction was purified into 30uL of water. The NanoDrop measured that the purified, digested plasmid was at 145ng/uL.
The digested hOR1D2 insert and pET-21A plasmid were ligated using 450ng of insert and 145ng of plasmid (aiming for the recommended 3:1 ratio of insert:plasmid for ligations). Ligation reactions were run overnight at 16C and then used to transform DH5alpha E.coli that were plated on LB/Agar plates containing 100ug/mL ampicillin.
The transformation unexpectedly produced a single colony. After some discussion, the competent DH5alpha cells used for this transformation were identified as cells from a batch that may have been inadvertently thawed over the summer when the -80C freezer it was originally stored in was emptied for maintenance and cleaning. This thawing would have certainly compromised the competence of the cells. This single colony was used to inoculate an overnight culture and the plasmid was isolated by miniprep (50uL of 860ng/uL).
To verify ligation of the hOR1D2 insert into pET-21A, the isolated plasmid was digested by EcoRI and HindIII for 3 hours before being analyzed on an agarose gel. If the plasmid is correctly constructed, this digest will generate a fragment that are ~950bp and ~5.4kb (Figure 3A). The gel confirmed production of a ~950bp fragment, but the second fragment was at ~3.5kb instead of the anticipated ~5.4kb (Figure 3C). A second digest with NdeI was expected to generate fragment of ~400bp and ~6kb (Figure 3B). This NdeI digest, however, produced bands at ~700bp and ~4kb. At the time of the iGEM Wiki freeze, the discrepancies for these differences in the expected molecular weights of the digests was still being discussed and examined. Since the PCR reaction amplifying the hOR1D2 coding sequence worked as anticipated, we concentrate on the whether there is an issue with the plasmid (e.g.: perhaps a mislabeled stock was used?) and it is expected that the ligation reactions will be redone.
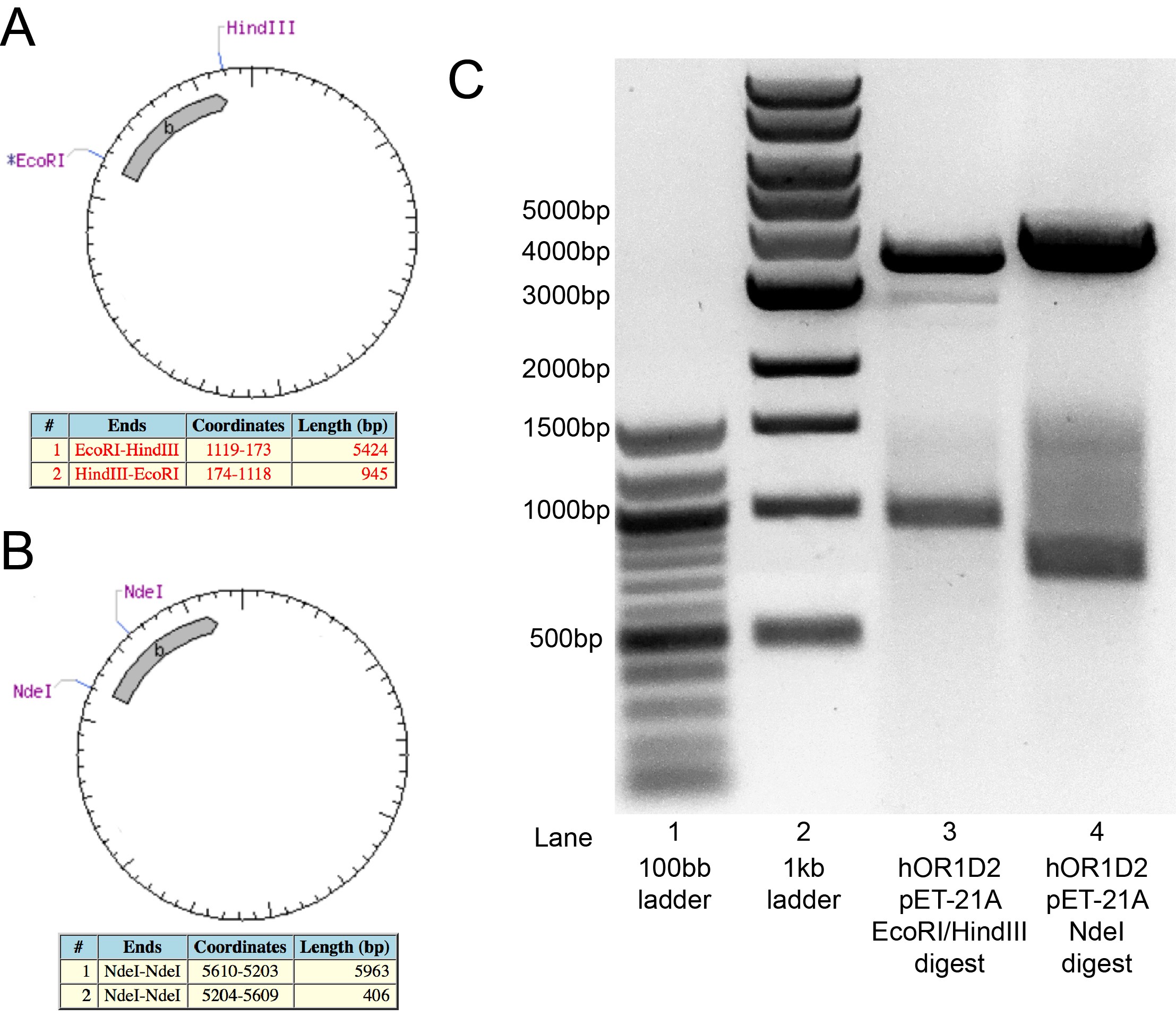
Figure 3: Analysis of hOR1D2 in pET-21A clone.
Construction of synthetic operon plasmid
An oligonucleotide was designed for generating the synthetic operon. This operon was designed to use the AraC-pBad promoter sequence that is identical to the AracC-Pbad promoter that is available in the iGEM parts database (part BBa_K808000). The coding regions for the His-tagged human Tau and Strep-tagged icaB genes were placed downstream of this promoter. Both genes were designed for E.coli codon use bias.
The oligonucleotide sequence for this synthetic operon was initially sent to IDT for its construction. The initial estimate of 4-6 weeks for its delivery, but after several months it was determined that the oligonucleotide could not be constructed as designed. An alternative vendor, GeneWiz, was contacted and offered to complete the construction and incorporate the operon into a pUC57 plasmid backbone. This construction was also reported several challenges that resulted in the multiple delays in the projected completion date. By the end of September, however, GeneWiz reported successful construction of a sequence-verified vector containing our synthetic operon.
Upon taking delivery of the synthetic operon plasmid, the plasmid was used to transform DH5alpha cells on LB/agar plates with 50ug/mL kanamycin. These colonies were used inoculate overnight growth generate a glycerol stock and plasmid stock (50uL at 900ug/uL). The plasmid has also been used to transform BL21(DE3) cells also on LB/agar plates with 50ug/mL kanamycin.
Future Directions
The BL21(DE3) cells transformed with the synthetic operon will be used to establish that the His-tagged human Tau and Strep-tagged icaB genes can be induced with arabinose. These experiments will grow cells on minimal media with glycerol as a carbon source, which should suppress expression of the operon genes. When the carbon source is switched to arabinose, however, the expression of the operon genes should be induced. This expression will be verified by qPCR and western blotting.
Once the construction issues for hOR1D2 in pET-21A have been resolved, this plasmid will be used with the synthetic operon plasmid to co-transform BL21(DE3) cells. These co-transformed cells will be used to test whether both the operon and hOR1D2 genes can be simultaneous co-expressed. This experiment be performed in the same way as the operon-only studies (see above), but in addition to arabinose, IPTG will also be added since this will be required to drive expression from the pET-21A plasmid. The co-expression of the operon and OR1D2 genes will also be verified by qPCR and western blotting.