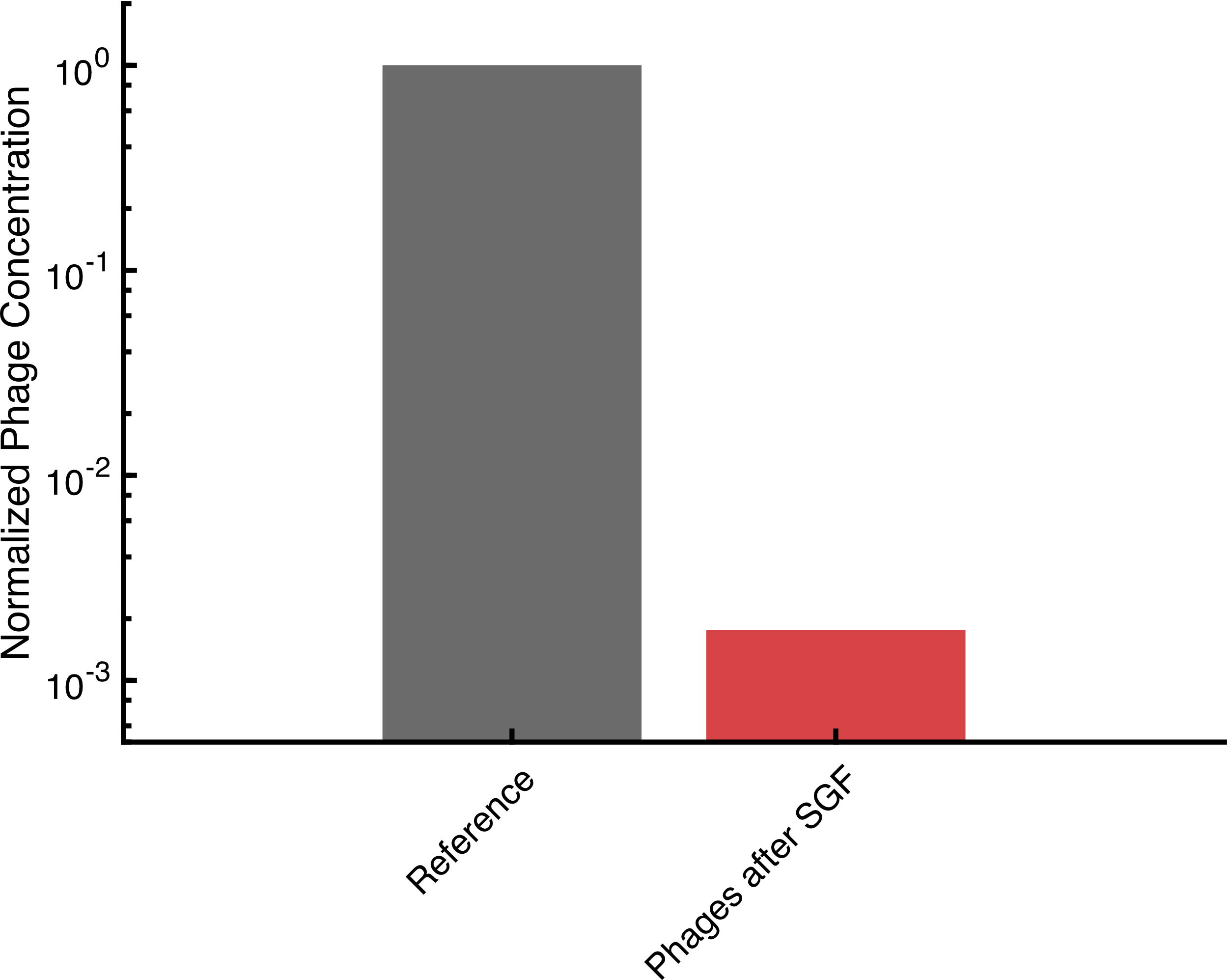|
|
| Line 254: |
Line 254: |
| | </div> | | </div> |
| | </div> | | </div> |
| | + | |
| | + | div class="row"> |
| | + | <div class="col-12"> |
| | + | <p> |
| | + | The TX-TL systems performed differentially regarding phage titers (E10>myTXTL>P15), protein content (E10>P15>myTXTL) and protein expression (P15>E10>Arbor), the proteome of the samples E10, P15 and myTXTL were analyzed by shotgun sequencing. |
| | + | The main question was, if the difference, especially in phage titers, is due to a different composition of the cell extracts or caused by a discrepancy in activity of the relevant proteins e.g. transcription and translation related proteins. |
| | + | Therefore, in collaboration with the Bavarian Biomolecular Mass Spectrometry Centre (BayBioMS) the samples were analyzed under supervision of Dr. Christina Ludwig. |
| | + | |
| | + | HEATMAP |
| | + | Heatmap of the label-free quantification intensities of all proteins identified. Diagram shows duplicate samples from the three cell extracts analyzed. Green indicates high expression, red indicates low expression. |
| | + | </p> |
| | + | |
| | + | <p> |
| | + | The results of the Mass Spectrometry gave us insight into the composition of the different TX-TLs. We were able to identify 1771 proteins in all the extracts. The results indicate a high homogenicity between all three TX-TLs, as illustrated in the heatmap. |
| | + | Based on the LFQ values (Label Free Quantification) a volcano plot of two samples (Arbor/E10, Arbor/P15 and E10/P15) was generated. </p> |
| | + | <p> |
| | + | VOLCANO |
| | + | In general, there is no protein more frequent in Arbor TX-TL in comparison to E10 and P15. In contrast, some translation-related proteins were slightly more abundant in E10 and P15, for instance certain tRNA ligases (PheS and PheT). The RecBCD subunits (the dsDNA degrading complex) are of similar abundance in all TX-TLs, showing the importance to consider its negative impact on the assembly. The results were further validated with DAVID, a tool for finding metabolic pathways based on proteomic data. The identified pathways indicate no correlation with transcription/translation related pathways (data not shown). |
| | + | </p> |
| | + | |
| | + | <p> |
| | + | In conclusion, the performance differences are most likely caused by a loss of activity during TX-TL preparation rather than changes in composition. |
| | + | </p> |
| | + | |
| | + | </div> |
| | + | </div> |
| | + | |
| | + | <hr> |
| | | | |
| | <div class="row"> | | <div class="row"> |
| | <div class="col-12 col-md-6"> | | <div class="col-12 col-md-6"> |
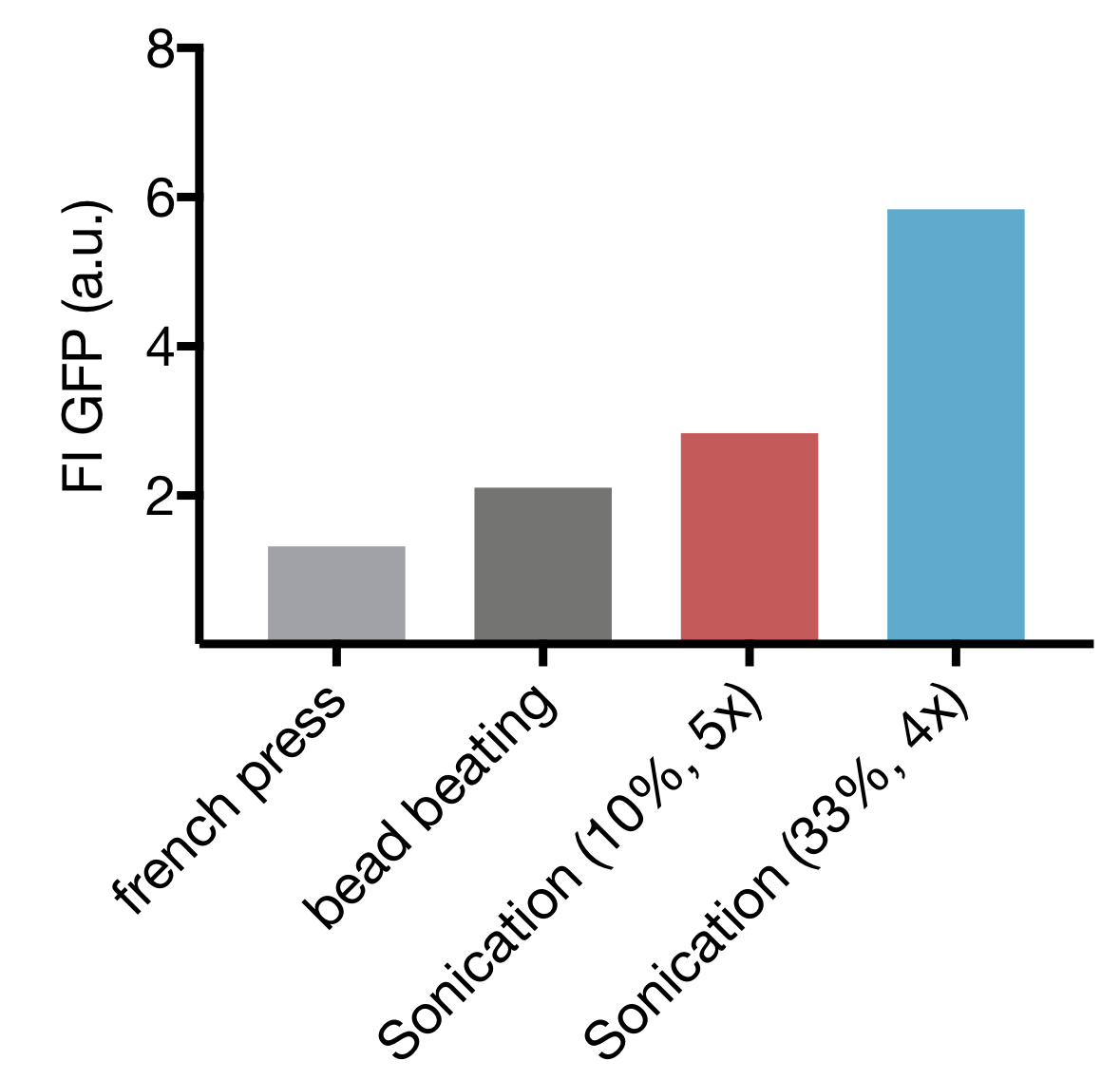
| − | <p>Discussion goes here | + | <p>The DNA sequence added to the cell-free system serves as the template for the required phage. Therefore, it is translated to RNA by an RNA-Polymerase. In addition, DNA-Polymerases can amplify the DNA segment, multiplying the amount of DNA in the cell-free reaction. |
| | + | To assess this effect and its dependence on deoxynucleotide triphosphates (dNTPs), we performed an absolute quantification of T7 DNA in the cell-free reaction by quantitative PCR (qPCR). A standard curve with a serial dilution of T7 DNA. We used the TXTL qPCR protocol (add link). |
| | + | As a reference, we used the myTXTL (Arbor Biosciences) cell-free reaction system. The addition of dNTP to the reference reaction leads to an increase in DNA concentration by a factor of 15 in the reaction after 4 hours (290 ng compared to 19 ng). This is higher than in the myTXTL reaction without additional dNTPs, in which there is a 1.8-fold increase in DNA (91 ng compared to 51 ng) after the 4-hour reaction. |
| | + | The home-made cell extracts P10 and E15 however do not resemble this behavior. |
| | + | |
| | </p> | | </p> |
| | </div> | | </div> |
| Line 268: |
Line 300: |
| | </div> | | </div> |
| | | | |
| − | | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p> |
| | + | It would be desirable to increase DNA amplification in our cell extracts. We therefore conducted a cause analysis, focusing on the T7 replication system. A more than 250-fold increase in processivity of the T7 DNA polymerase is achieved by its binding behavior to E. coli thioredoxin. We suspected reduced presence of this factor in our cell extract. Thioredoxin could be added to a phage assembly reaction to further test these assumptions. However, our proteome analysis did not confirm that there were low levels of thioredoxin present in our cell extract. |
| | + | </p> |
| | + | |
| | + | </div> |
| | + | </div> |
| | | | |
| | | | |
| Line 318: |
Line 357: |
| | <div class="row"> | | <div class="row"> |
| | <div class="col-12 col-md-6"> | | <div class="col-12 col-md-6"> |
| − | <p>eeeh | + | |
| − | </p> | + | |
| | + | <p>Endotoxins are pyrogens deriving from gram-negative bacteria. Their mini from any pharmaceutical product is mandatory. Therefore, or Phactory, we engineered an E. coli strain lacking lipid A, a major endotoxin component and used this bacterium to produce our cell extract To evaluate endotoxin content of different cell extracts, a Limulus Amebocyte Lysate (LAL)-test was performed according to the <a href="https://www.genscript.com/product/documents?cat_no=L00350&catalogtype=Document-PROTOCOL">supplier manual</a>. As a reference, we compared the cell extract from our msbB-deficient strain (K2) to a cell extract from a wild-type strain (K4) as well as a commercial cell-free system (myTXTL, Arbor Biosciences). A solution with live <i>E. coli</i> served as a positive control.</p> |
| | + | <p>Compared to the K4 strain our msbB-deficient K2 cell extract had 49-fold reduced endotoxin levels (0.06 EU/ml compared to 2.94 EU/ml). Other cell extracts such as the P15 cell extract (3.83 EU/ml) and the commercial myTXTL (4.65 EU/ml) had even higher endotoxin contents.</p> |
| | + | |
| | + | |
| | + | |
| | </div> | | </div> |
| | <div class="col-12 col-md-6"> | | <div class="col-12 col-md-6"> |
| | <figure class="figure"> | | <figure class="figure"> |
| | <img src="https://static.igem.org/mediawiki/2018/5/53/T--Munich--Results_Wl2_LAL.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | | <img src="https://static.igem.org/mediawiki/2018/5/53/T--Munich--Results_Wl2_LAL.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> |
| − | <figcaption class="figure-caption">Our two tested samples of cell extract retained 70 and 90 % of expression quality respectively after lyophilization.</figcaption> | + | <figcaption class="figure-caption">Endotoxin content in different cell-extracts determined by LAL-Test. Error bars indicate standard deviation of the measured plateau values. Error bars indicate SD.</figcaption> |
| | </figure> | | </figure> |
| | </div> | | </div> |
| | </div> | | </div> |
| | | | |
| | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p>A calibration curve using known endotoxin concentrations is required for the LAL-Test. A dilution series ranging from 0.625 EU/ml to 5 EU/ml. The fitting curve is used to interpolate the concentrations in the unknown sample. The linear fit of the calibration curve had a R2 of 0.98, an intersection with the y-axis at 0.38 and a slope of 0.39 ml/EU. These values are in accordance with the requirements of the LAL-Test manufacturer. (Supplemental Data)</p> |
| | + | </div> |
| | + | </div> |
| | + | |
| | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p>Removal of endotoxins is impeded by their tendency to form stable interactions with other biomolecules. Our method of preventing the lipid A biosynthesis is therefore superior to extensive isolation steps required for removing endotoxins in conventional phage production. </p> |
| | + | </div> |
| | + | </div> |
| | + | |
| | <hr> | | <hr> |
| | | | |
| Line 359: |
Line 415: |
| | Cell-Free Systems Allow Host Independent Bacteriophage Assembly | | Cell-Free Systems Allow Host Independent Bacteriophage Assembly |
| | </h3> | | </h3> |
| | + | |
| | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p> |
| | + | Phactory has is the ability to assemble any bacteriophage, in a host-independent manner. To underline this feature and demonstrate universal applicability, we assembled a variety of different E. coli phages, both DNA and RNA-based. |
| | + | </p> |
| | + | <br> |
| | + | <br> |
| | + | </div> |
| | + | </div> |
| | + | |
| | + | <div class="row"> |
| | + | <div class="col-12 col-md-6"> |
| | + | <p>The successful assembly of all phages was confirmed by plaque assay and transmission electron microscopy (TEM). In addition, DNA encoding for NES and FFP phages was used to perform assembly of these phages in our cell extract. However, we were not in possession of the respective host bacterial strains and therefore could not demonstrate successful assembly. |
| | + | </p> |
| | + | |
| | + | </div> |
| | + | <div class="col-12 col-md-6"> |
| | + | <figure class="figure"> |
| | + | <img src=" https://static.igem.org/mediawiki/2018/7/76/T--Munich--Results--WL3_Titer_difference.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> |
| | + | <figcaption class="figure-caption">titer_dif</figcaption> |
| | + | </figure> |
| | + | </div> |
| | + | </div> |
| | + | |
| | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p> |
| | + | Pathogenic bacteria such as salmonella, pseudomonas and staphylococcus are prone to develop multi-drug resistance and pose an urgent or serious threat (Centers for Disease Control and Prevention, 2013. Antibiotic/Antimicrobial Resistance.). Therefore, to fulfill this medical need, bacteriophages specific for these bacteria should be assembled next in our cell-free system. |
| | + | </p> |
| | + | <br> |
| | + | <br> |
| | + | </div> |
| | + | </div> |
| | + | |
| | <hr> | | <hr> |
| | <h3> | | <h3> |
| | Bacteriophages From Phactory Assemble And Are Fully Functional | | Bacteriophages From Phactory Assemble And Are Fully Functional |
| | </h3> | | </h3> |
| − | | + | |
| | + | |
| | <div class="row"> | | <div class="row"> |
| | <div class="col-12 col-md-6"> | | <div class="col-12 col-md-6"> |
| Line 376: |
Line 468: |
| | </div> | | </div> |
| | </div> | | </div> |
| − | | + | |
| | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p> |
| | + | To rapidly detect functionality of the phages, Reverse Transcription-quantitative PCR (RT-qPCR) was applied to pellets of T7-infected host cells at different timepoints. The RT-qPCR protocol was used (add link). Using the delta-delta-Ct method3, relative expression was determined and normalized to the value at 3 minutes after addition of the phages. Whereas expression of E. coli genes remains stable, expression of three T7 genes is elevated throughout the experiment. The change of expression of all three phage genes reaches a first peak after 12 minutes and a second peak after 18 or 21 minutes. |
| | + | </p> |
| | + | <p> |
| | + | The sharp increase of phage gene expression in the first phase of the experiment displays the ability of the phages to successfully infect the bacteria and initiate reproduction. The second and even higher increase of expression is likely attributed to a second wave of infection of already replicated phages. This indicates that the phages are capable of reproducing inside their host bacteria, resulting in multiplication of functional phages. |
| | + | The increase of expression of T7P01 is more pronounced than that of T7P07, which is in turn is stronger than that of T7P29. This circumstance is likely caused by differences in primer efficacy, a value describing the doubling rate in between every PCR cycle. For better reliability of the RT-qPCR quality control, primer efficacy should be assessed by creating a standard curve. |
| | + | </p> |
| | + | <p>All RNA samples were pooled and checked for DNA contamination, which could have interfered with the RT-qPCR. Absence of DNA was determined electrophoresis in a gel electrophoresis</p> |
| | + | <br> |
| | + | <br> |
| | + | </div> |
| | + | </div> |
| | + | |
| | | | |
| | <div class="row"> | | <div class="row"> |
| | <div class="col-12 col-md-6"> | | <div class="col-12 col-md-6"> |
| − | <p>Discussion goes here | + | <p>To prove the influence of the DNA concertation on the bacteriophage titer, cell extract reaction were prepared with varying T4 DNA concentration. The bacteriophage production was performed according to the protocol (link). The titer of the bacteriophages was measured with the top agar method and the formed plaques were counted. The increase in DNA concentration results also in an increase in the bacteriophage concentration. This increase in nonlinear and our model predicted. |
| | </p> | | </p> |
| | </div> | | </div> |
| Line 403: |
Line 510: |
| | <hr> | | <hr> |
| | | | |
| | + | <div class="row"> |
| | + | <div class="col-12"> |
| | + | <p>We performed a Plaque Assay to determine the titer of viable phages in our assembly batch. By creating serial dilutions, we were able to calculate a plaque forming units/milliliter (PFU/ml) value. The plaque assay protocol (link) was used. |
| | + | </p> |
| | + | </div> |
| | + | </div> |
| | + | |
| | + | |
| | <div class="row"> | | <div class="row"> |
| | <div class="col-12 col-md-12"> | | <div class="col-12 col-md-12"> |
| Line 424: |
Line 539: |
| | </h3> | | </h3> |
| | | | |
| − | <div class="row">
| + | |
| − | <div class="col-12 col-md-6">
| + | |
| − | <p>Discussion goes here
| + | |
| − | </p>
| + | |
| − | </div>
| + | |
| − | <div class="col-12 col-md-6">
| + | |
| − | <figure class="figure">
| + | |
| − | <img src=" https://static.igem.org/mediawiki/2018/7/76/T--Munich--Results--WL3_Titer_difference.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure.">
| + | |
| − | <figcaption class="figure-caption">dna conc</figcaption>
| + | |
| − | </figure>
| + | |
| − | </div>
| + | |
| − | </div>
| + | |
| − |
| + | |
| | | | |
| | | | |
We sequenced several phage genomes after preparation using ….
After receiving the phage genomes, one of the first steps to do is analyse the received sequences.
This has been done using an inhouse software poreSTAT [1].
For each sequencing sample, it is analysed how many bases are sequenced, what bp yield has been achieved and how many pore were used.
Considering the sequence in which the reads have been acquired, it can nicely be seen how the used chip gets worn out with each sequencing experiment.
While for the T7 sequencing almost all pores are usable, the used pores decreases with every sequencing experiment …
_*_*
4 missing figures
_*_*
While the T7 experiment produced most reads and most base-pairs sequenced, the remaining three experiments produced less data.
The read and base-pair yield can be seen in Table 1.
TABLE
The actual task here was to assemble the phage genomes from the Nanopore reads.
Particularly Nanopore sequencing is well suitable for genome assembly, since its long reads allow to reduce ambiguity of highly similar and repetitive sequences.
Figure 2 shows the reads distributions of the sequencing experiments.
_*_*
4 missing figures
_*_*
However, during the initial screening it could be seen, that the read lengths do not approach the thought length of the phage genomes in the 50-70kbp range for T7 and 100 kbp range for the remaining phages.
Smaller reads generally lead to more ambiguity and thus are to be avoided from a bioinformatics point of view.
However, since no bioinformatician can change the sequenced data afterwards, we went on with the given data.
There are several tools available for de-novo genome assembly in general, however most approaches are used for short-read genome assembly (2nd generation sequencing, Illumina) and employ a de bruijn graph approach.
Here we have 3rd generation sequencing data which must be handled totally different from old short-read sequencing data: the reads are less perfect in terms of sequencing errors. While short-reads nowadays have error-rates of about 1% (e.g. 1 base out of 100 is reported incorrectly), this error is up to 15% for nanopore sequencing data using newest sequencing chemistry (R9.4 at the time of wiki-freeze).
Thus the number of available assemblers drops dramatically, where canu [2] and miniasm [3] are the most prevalent ones. Both rely on a overlap-layout-consensus approach, which, historically, can be seen as the father of all assemblers (see celera assembler [5]).
We first used canu to assemble our genomes. Unfortunately we have been confronted with a major problem: contamination. We thus developed sequ-into to first detect the contamination and also get rid of contamination-originated reads.
More on the performance and finding while using sequ-into can be found at …[link to /Software].
Particularly for assembly, contamination is bad because it can lead the assembler into wrong directions – depending on the phylogenetic distance of the original sample and the contamination.
After getting rid of the contamination we noticed some strange patterns in the phage genome assemblies after re-aligning the reads to the assembly, which can be seen in Figure 3.
*_*_
missing figure
*_*_
In theory we can expect a uniform coverage over the full genome, since there is no bias for read template generation during sample preparation (thanks to random primers).
However, what we saw here is that the first part has lower coverage than the remaining part and there is a high-coverage region in the middle of the assembled genome.
Since we know phage genomes may have repitions, we thought to splite the genome in the middle and reorganise the structure to no avail.
We thus tried to use the other assembler, miniasm, which is known for very fast assemblies, with little error correction.
However, this error correction can be achieved by combining miniasm with minimap for read mapping and racon for polishing the sequences.
Thus, the assembly pipeline changed to the following calls:
MAY BE COLLAPSIBLE
#!/usr/bin/env sh
INREADS=$1
ASMFOLDER=$2
ASMPREFIX=$3
THREADS=$4
if [ -z "$4" ]
then
THREADS=4
fi
# path to used executables
MINIMAP2=minimap2
MINIASM=miniasm
GRAPHMAP=graphmap
RACON=racon
# first we must overlap all reads with each other
$MINIMAP2 -x ava-ont -t$THREADS $INREADS $INREADS > $ASMFOLDER/$ASMPREFIX.paf
# then miniasm can create alignment
$MINIASM -f $INREADS $ASMFOLDER/$ASMPREFIX.paf > $ASMFOLDER/$ASMPREFIX.gfa
# extract unitigs from miniasm
awk '$1 ~/S/ {print ">"$2"\n"$3}' $ASMFOLDER/$ASMPREFIX.gfa > $ASMFOLDER/$ASMPREFIX.unitigs.fasta
# align reads with unitigs
$MINIMAP2 $ASMFOLDER/$ASMPREFIX.unitigs.fasta $INREADS > $ASMFOLDER/$ASMPREFIX.unitigs.paf
# find contigs from unitigs
$RACON $INREADS $ASMFOLDER/$ASMPREFIX.unitigs.paf $ASMFOLDER/$ASMPREFIX.unitigs.fasta > $ASMFOLDER/$ASMPREFIX.contigs.fasta
~/progs/minimap2/minimap2 -x map-ont -a -t$THREADS $ASMFOLDER/$ASMPREFIX.contigs.fasta $INREADS > $ASMFOLDER/$ASMPREFIX.reads.mm2.sam
$GRAPHMAP align -r $ASMFOLDER/$ASMPREFIX.contigs.fasta -d $INREADS -o $ASMFOLDER/$ASMPREFIX.reads.gm.sam
And can be started simply from the command-line using: ./assemble.sh "FQ file" "PATH TO ASM FOLDER" "PREFIX of output"
This finally led to a good assembly after rearranging the middle part which initially was “over-expressed”.