
Optimizing a Cell-Free Expression System
The first step of our project is the optimization of a cell extract as a manufacturing platform for bacteriophages. For this purpose, it was necessary to produce a high-quality cell extrac, in a reproducible and easy manner. We found that
- biorector cultivation, sonication, lysozyme enhance the performance
- different cultivation and cell disruption methods do not overly impact the protein composition, but probably the activity of the expressed protein
- lyophylization is a good way to store cell extract
- the run-off reaction as suggested by Sun et al. is indeed beneficial and dialysis is superior to diafiltration via centrifugal filter
Bioreactors Allow for Upscaling of Cell Extract Production
Cultivation is the first step in cell extract preparation. The original cell extract preparation protocol uses shaking flask cultivation for biomass production and states that cell harvest at OD 1,8-2,0 is strictly required to produce high-quality extract. To be able to transfer this to the bioreactor we first obtained growth data for both shaking flask and bioreactor cultivation.

The growth curve from shaking flask cultivation showed us that harvest at OD 1,8-2 correlates to the mid-to late logarithmic growth phase. In bioreactor fermentation this correlates to OD between 4 and 6. We decided to test which of these ODs is best to harvest cells for cell extract preparation.

This experiment gave 2 important results:
- the optimal OD to harvest culture from the bioreactor is around 5. This gives the highest protein content and also the best expression.
- preparing cell extract from bioreactor cultivated cells under comparable conditions gives equal quality extract than cell extract from shaking flask cultivated cells.
In our small lab-scale bioreactor with 2 L cultivation volume we were able to obtain 20 g cell pellet. 2 L cultivation in shaking flasks only yields 4,5 g pellet.
Sonication and Lysozyme Improve the Performance of Cell-Extract
Sonication
Of the three commonly used methods of cell lysis, each has their advantages and drawbacks. For cell extract preparation bead beating is most established, but the caveat there is, that it doesn’t leave room for upscaling, besides being tedious and time-consuming. Less often used is high-pressure cell disruption in a so called ‘french-press’, but these devices are very expensive and not widely prevalent which impedes our effort to provide a generally-applicable protocol for preparation of home-made cell extract. A sonication device on the other hand is available in most biochemical labs as it’s a widely used method for cell lysis in protein purification protocols. Nevertheless, we started by comparing all 3 lysis methods with settings commonly applied for cell lysis. For Sonication two commonly used sets of parameters were used.
Our Initial test showed that cell lysis by Sonication yields extract with up to 2,8 and 4,4 fold higher expression than cell lysis by bead beating and French press respectively. These findings, combined with the limited options of optimization for both bead beating and French press lysis as well as restricted potential for upscaling of bead beating lead to the decision to focus our optimization efforts on Sonication as Lysis method.
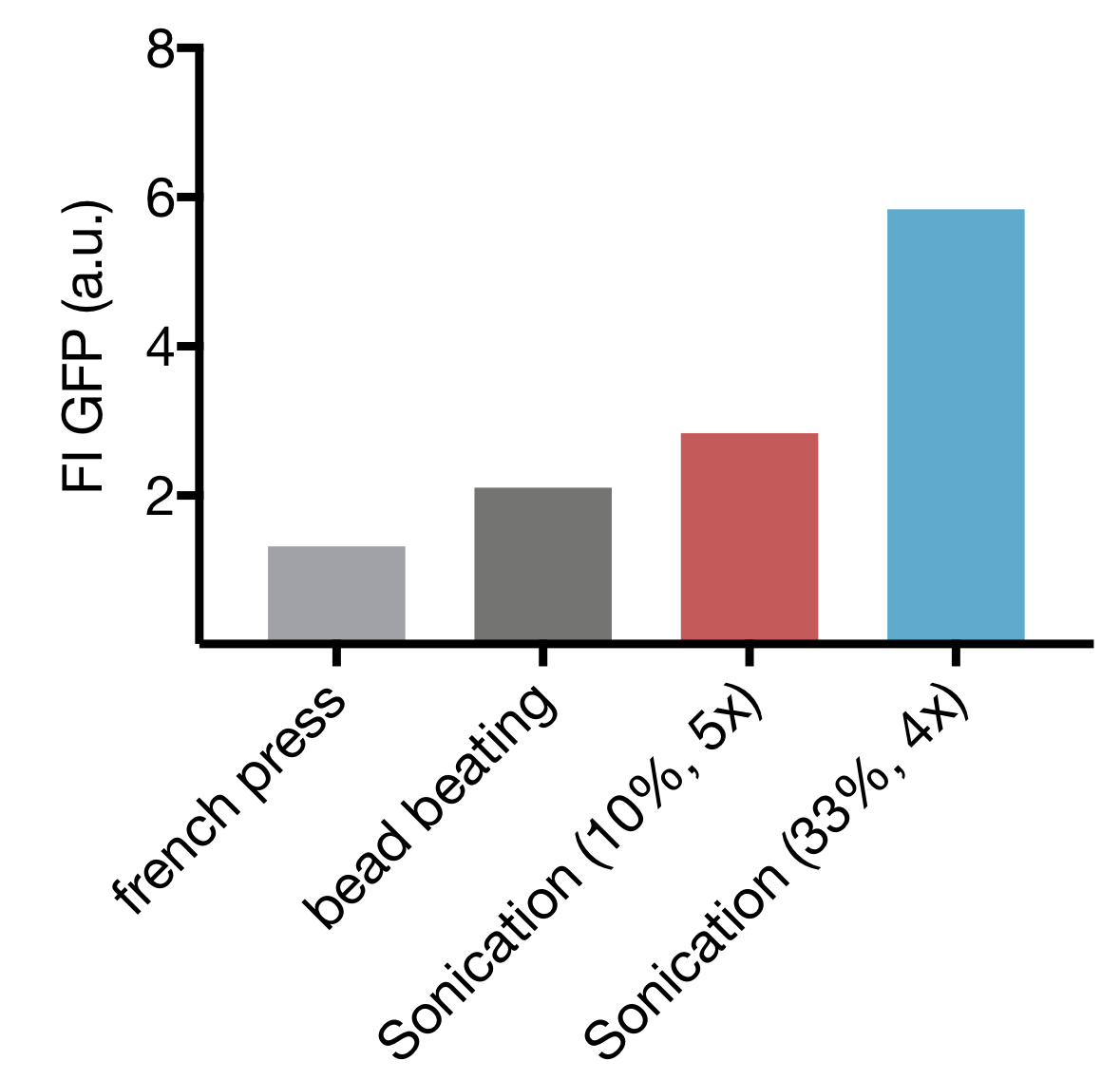
Comparison of different cell Lysis methods: Sonication at 33 % Amplitude, 4 cycles; Sonication at 10 % Amplitude, 5 cycles; bead beating and high-pressure disruption (french press) – Expression Quality in cell extract measured as GFP Fluorescence Intensity
Cell Lysis by Sonication offers 2 main parameters that can be varied: the Amplitude, that correlates to the energy emited by the device used and Cycle number, representing the total time of Sonication. AS mentioned in XY paper we decided to use 15 s pulses with 10 s pause between pulses to prevent excessive heating of the sample.
Our screening revealed that cycle numbers above 33 % seem to be inept for cell extract preparation, therefore our further efforts focused on Lysis with 10 % and 33 % amplitudes. Smaller cycle numbers offer the additional benefit of shorter overall processing time, cutting the cell expenditure of time for cell extract preparation short.

Lysozyme
In the last step we made an effort towards upscaling of the sonication step. Preliminary experiments (results not shown) indicated 4 mL as the maximal sample volume that could efficiently be lysed with the device we had at our disposal. As indicated in the XY paper we decided to increase the number of sonication cycles in proportion to the increase of sample volume. The presented test revealed that this approach was successful; protein content in the cell extract and expression quality was not ustr maintained but increased through the use of higher sample volumes. This proves that upscaling of cell extract preparation with cell lysis by sonication is indeed feasible.


Additionally we tested the effect of addition of the cell-wall degrading enzyme Lysozyme to the cell lysis, following a tip from a lab member. To our knowledge usage of Lysozyme in cell extract preparation has not been a focus of previous studies. All the more surprised were we, to find that Lysozyme increases the protein yield of cell lysis for all tested settings. Moreover, the expression quality was likewise increased after Lysozyme addition, indicating that the remaining enzyme does not interfere with protein biosynthesis.
Cultivation and Extract Preparation Barely Impacts Cell Extract Composition
The main question was, if the different performance of the cell extracts (P15, E10, myTXTL), especially concerning phage titers, is due to a divergent composition or caused by a discrepancy in activity of the relevant proteins e.g. transcription and translation related proteins Therefore, one student was sent to Bavarian Biomolecular Mass Spectrometry Centre (BayBioMS), where he analyzed the samples under the supervision of Dr. Christina Ludwig.
The results of the Mass Spectrometry gave us insight into the composition of the different TX-TLs. We were able to identify 1771 proteins in all the extracts. The results indicate a high homogeneity between all three TX-TLs, as illustrated in the heatmap. Based on the LFQ values (Label Free Quantification) a volcano plot of two samples (Arbor/E10, Arbor/P15 and E10/P15) was generated. In general, there is no protein more frequent in Arbor TX-TL in comparison to E10 and P15. In contrast, some translation related proteins were slightly more abundant in E10 and P15 like certain tRNA ligases (PheS and PheT). The RecBCD subunits (the dsDNA degrading complex) is of similar abundance in all TX-TLs, showing the importance to consider its negative impact on the assembly. The results were further validated with DAVID, a tool for finding metabolic pathways based on proteomic data. The identified pathways indicate no correlation with transcription/translation related pathways (data not shown).
We concluded that the performance difference is most likely due to decreased activity during TX-TL preparation rather than a change in composition.




Lyophilization Is a Good Choice for Cell-Extract Storage
For long term storage of our cell-extract we decided to try lyophilization. After initial tests we found that:
- cell extract quality is only preserved when cell-extract is mixed with the txtl- reaction buffer prior to lyophilization.
- the retention of quality does not depend on the size of lyophilized extract aliquots.
- We then tested the expression quality of several of our home-made cell extracts from fresh vs. lyophilized aliquots.
eeeh

Our two tested samples of cell extract retained 70 and 90 % of expression quality respectively after lyophilization.
Synthetic Phage Manufacturing
Phages were manufactured based on three components: the cell extract, an energy solution (ATP, GTP, NAD+ etc.) and a supplement solution (aminoacids, tRNAs, pholic acid etc.). The only additional component for phage assembly is pure phage DNA. We expanded the assembly platform by
- producing the first clinically relevant phages at therapeutic concentrations
- manufacturing of therapeutic phages independent of a living pathogen
- achieving similar phage titers as the commercial cell extract
- determining the amount of DNA necessary for sufficient phage assembly
- calculating the amount of DNA produced in the cell extract by replication
Home-Made Cell Extract Achieves Phage Titer Comparable to the Commercial System
We determined the concentration of phages assembled in our cell extracts P15 and E10, which had the most promising results in our cell extract quality control. We compared our extracts to the commercial extract (myTXTL) and saw a similar performance.
All in all our extract has the same phage assembly potential as the commercial cell extract. Moreover, lyophylization, which makes the cell extract more long lived, does not reduce the effiency of bacteriophage assembly. We concluded that phages could be assembled on site in a durable lyophilized cell extract.

Bacteriophages Can Be Assembled Independent of the Living Host
Phactory has is the ability to assemble various bacteriophages, in a bacteria-independent manner. To underline this feature and demonstrate universal applicability, we assembled a variety of different E. coli phages, both DNA and RNA-based.
The successful assembly of all phages was confirmed by plaque assay and transmission electron microscopy (TEM). In addition, DNA encoding for NES and FFP phages was used to perform assembly of these phages in our cell extract. However, we were not in possession of the respective host bacterial strains and therefore could not demonstrate successful assembly.
Pathogenic bacteria such as salmonella, pseudomonas and staphylococcus are prone to develop multi-drug resistance and pose an urgent or serious threat (Centers for Disease Control and Prevention, 2013. Antibiotic/Antimicrobial Resistance.). Therefore, to fulfill this medical need, phages specific for these bacteria should be assembled next in our cell-free system. Potentially, this would require co-expression of the respective sigma factors that are needed for initiation of transcription.







DNA Concentration Determines Phage Titer
Bacteriophage Titers Correlate With DNA Concentration
To prove the influence of the DNA concentration on the bacteriophage titer, cell extract reactions were prepared with varying T4 DNA concentrations. The bacteriophage production was performed. The titer of the bacteriophages was measured with the top agar method and the formed plaques were counted. The increase in DNA concentration results also in an increase in the bacteriophage concentration. This increase is nonlinear and our model predicted. This finding is probably due to a critical concentration of phage proteins, which have to be reached for capsid head assembly (similar to a critical micelle concentration).


Cell-Free Systems Replicate Phage Genomes
The DNA sequence added to the cell-free system serves as the template for the required phage. We saw, that DNA-Polymerases can amplify the DNA segment, multiplying the amount of DNA in the cell-free reaction.
To assess this effect and its dependence on deoxynucleotide triphosphates (dNTPs), we performed an absolute quantification of T7 DNA in the cell-free reaction by quantitative PCR (qPCR). A standard curve with a serial dilution of T7 DNA. We used the TXTL qPCR protocol (add link). We used two selected cell extracts from us (P15 and E10), which reached similar phage titers compared to the commercial cell extract (myTXTL) in previous experiments. We compared the replication potential in comparison to myTXTL.
The addition of dNTP to the reference reaction leads to an increase in DNA concentration by a factor of 15 in the reaction after 4 hours (290 ng compared to 19 ng). This is higher than in the myTXTL reaction without additional dNTPs, in which there is a 1.8-fold increase in DNA (91 ng compared to 51 ng) after the 4-hour reaction. The home-made cell extracts P10 and E15 however do not resemble this behavior.
It would be desirable to increase DNA amplification in our cell extracts. We therefore conducted a cause analysis, focusing on the T7 replication system. A more than 250-fold increase in processivity of the T7 DNA polymerase is achieved by its binding behavior to E. coli thioredoxin1. We suspected reduced presence of this factor in our cell extract. Thioredoxin could be added to a phage assembly reaction to further test these assumptions. However, our proteome analysis did not confirm that there were low levels of thioredoxin present in our cell extract.

Modelling Phage Production in Cell Extract
Modular Bacteriophage Composition
In the TX-TL system should be possible to modify phage proteins without altering their genome. This was attempted by modifying HOC (highly immunogenic capsid protein), which is part of the capsid protein structure of the T4-phage. Therefore, His-TEV-YFP-HOC was separately expressed and the purified protein was applied to our phage assembly.
For protein modification of the T4 capsid protein HOC, a plasmid for protein expression was cloned and His-YFP-HOC (82kDa) was expressed. The plasmid was transformed into BL21, expressed and puryfied by nickel affinity chromatography and gelfiltration.
FILM
MODEL

After the two chromatography steps, the purified protein had a final concentration of 70µM and no by-products were visible by silver staining. The CD-spectra (minima of 218nm) of the protein corresponds to the model and the Ramachandran plot indicates that the protein was not denatured during purification. Then, the purified protein was intentionally denatured by thermal transition. Thereby, it was confirmed that the protein is stable below 40°C. This is of importance, as phage assembly is performed at 29°C.



The purified protein was added to the assembly mix. Bacteria were transfected with the modified phages. Unbound phages and protein were removed by centrifugation. Fluorescence was measured in dependence of the proximity to the bacteria. Theoretically, YFP intensity should correlate with the binding of YFP-HOC modified phages to the bacteria.


The purified protein was added to the assembly mix. Bacteria were transfected with the modified phages. Unbound phages and protein were removed by centrifugation. Fluorescence was measured in dependence of the proximity to the bacteria. Theoretically, YFP intensity should correlate with the binding of YFP-HOC modified phages to the bacteria.
Quality Control
Assessing Phage Functionality by Plaque Assays
We performed a Plaque Assay to determine the titer of viable phages in our assembly batch. By creating serial dilutions, we were able to calculate a plaque forming units/milliliter (PFU/ml) value. The plaque assay protocol (link) was used.


Assessing Endotoxin Levels
Msb-B Knockouts Reduce Endotoxin Levels By 49-Fold
Endotoxins are pyrogens deriving from gram-negative bacteria. Their mini from any pharmaceutical product is mandatory. Therefore, or Phactory, we engineered an E. coli strain lacking lipid A, a major endotoxin component and used this bacterium to produce our cell extract To evaluate endotoxin content of different cell extracts, a Limulus Amebocyte Lysate (LAL)-test was performed according to the supplier manual. As a reference, we compared the cell extract from our msbB-deficient strain (K2) to a cell extract from a wild-type strain (K4) as well as a commercial cell-free system (myTXTL, Arbor Biosciences). A solution with live E. coli served as a positive control.
Compared to the K4 strain our msbB-deficient K2 cell extract had 49-fold reduced endotoxin levels (0.06 EU/ml compared to 2.94 EU/ml). Other cell extracts such as the P15 cell extract (3.83 EU/ml) and the commercial myTXTL (4.65 EU/ml) had even higher endotoxin contents.

A calibration curve using known endotoxin concentrations is required for the LAL-Test. A dilution series ranging from 0.625 EU/ml to 5 EU/ml. The fitting curve is used to interpolate the concentrations in the unknown sample. The linear fit of the calibration curve had a R2 of 0.98, an intersection with the y-axis at 0.38 and a slope of 0.39 ml/EU. These values are in accordance with the requirements of the LAL-Test manufacturer.
Removal of endotoxins is impeded by their tendency to form stable interactions with other biomolecules2. Our method of preventing the lipid A biosynthesis is therefore superior to extensive isolation steps required for removing endotoxins in conventional phage production.
Sequ-Into Allows For Accurate Quantification of DNA Contamination Levels
We sequenced several phage genomes after preparation using …. After receiving the phage genomes, one of the first steps to do is analyse the received sequences. This has been done using an inhouse software poreSTAT [1]. For each sequencing sample, it is analysed how many bases are sequenced, what bp yield has been achieved and how many pores were used. Considering the sequence in which the reads have been acquired, it can nicely be seen how the used chip gets worn out with each sequencing experiment. While for the T7 sequencing almost all pores are usable, the used pores decreases with every sequencing experiment … _*_* 4 missing figures _*_* While the T7 experiment produced most reads and most base-pairs sequenced, the remaining three experiments produced less data. The read and base-pair yield can be seen in Table 1.
TABLE
The actual task here was to assemble the phage genomes from the Nanopore reads. Particularly Nanopore sequencing is well suitable for genome assembly, since its long reads allow to reduce ambiguity of highly similar and repetitive sequences. Figure 2 shows the reads distributions of the sequencing experiments. _*_* 4 missing figures _*_* However, during the initial screening it could be seen, that the read lengths do not approach the thought length of the phage genomes in the 50-70kbp range for T7 and 100 kbp range for the remaining phages. Smaller reads generally lead to more ambiguity and thus are to be avoided from a bioinformatics point of view. However, since no bioinformatician can change the sequenced data afterwards, we went on with the given data. There are several tools available for de-novo genome assembly in general, however most approaches are used for short-read genome assembly (2nd generation sequencing, Illumina) and employ a de bruijn graph approach. Here we have 3rd generation sequencing data which must be handled totally different from old short-read sequencing data: the reads are less perfect in terms of sequencing errors. While short-reads nowadays have error-rates of about 1% (e.g. 1 base out of 100 is reported incorrectly), this error is up to 15% for nanopore sequencing data using newest sequencing chemistry (R9.4 at the time of wiki-freeze). Thus the number of available assemblers drops dramatically, where canu [2] and miniasm [3] are the most prevalent ones. Both rely on a overlap-layout-consensus approach, which, historically, can be seen as the father of all assemblers (see celera assembler [5]). We first used canu to assemble our genomes. Unfortunately we have been confronted with a major problem: contamination. We thus developed sequ-into to first detect the contamination and also get rid of contamination-originated reads. More on the performance and finding while using sequ-into can be found at …[link to /Software]. Particularly for assembly, contamination is bad because it can lead the assembler into wrong directions – depending on the phylogenetic distance of the original sample and the contamination. After getting rid of the contamination we noticed some strange patterns in the phage genome assemblies after re-aligning the reads to the assembly, which can be seen in Figure 3. *_*_ missing figure *_*_
In theory we can expect a uniform coverage over the full genome, since there is no bias for read template generation during sample preparation (thanks to random primers). However, what we saw here is that the first part has lower coverage than the remaining part and there is a high-coverage region in the middle of the assembled genome. Since we know phage genomes may have repitions, we thought to splite the genome in the middle and reorganise the structure to no avail. We thus tried to use the other assembler, miniasm, which is known for very fast assemblies, with little error correction. However, this error correction can be achieved by combining miniasm with minimap for read mapping and racon for polishing the sequences. Thus, the assembly pipeline changed to the following calls:
MAY BE COLLAPSIBLE #!/usr/bin/env sh INREADS=$1 ASMFOLDER=$2 ASMPREFIX=$3 THREADS=$4 if [ -z "$4" ] then THREADS=4 fi # path to used executables MINIMAP2=minimap2 MINIASM=miniasm GRAPHMAP=graphmap RACON=racon # first we must overlap all reads with each other $MINIMAP2 -x ava-ont -t$THREADS $INREADS $INREADS > $ASMFOLDER/$ASMPREFIX.paf # then miniasm can create alignment $MINIASM -f $INREADS $ASMFOLDER/$ASMPREFIX.paf > $ASMFOLDER/$ASMPREFIX.gfa # extract unitigs from miniasm awk '$1 ~/S/ {print ">"$2"\n"$3}' $ASMFOLDER/$ASMPREFIX.gfa > $ASMFOLDER/$ASMPREFIX.unitigs.fasta # align reads with unitigs $MINIMAP2 $ASMFOLDER/$ASMPREFIX.unitigs.fasta $INREADS > $ASMFOLDER/$ASMPREFIX.unitigs.paf # find contigs from unitigs $RACON $INREADS $ASMFOLDER/$ASMPREFIX.unitigs.paf $ASMFOLDER/$ASMPREFIX.unitigs.fasta > $ASMFOLDER/$ASMPREFIX.contigs.fasta ~/progs/minimap2/minimap2 -x map-ont -a -t$THREADS $ASMFOLDER/$ASMPREFIX.contigs.fasta $INREADS > $ASMFOLDER/$ASMPREFIX.reads.mm2.sam $GRAPHMAP align -r $ASMFOLDER/$ASMPREFIX.contigs.fasta -d $INREADS -o $ASMFOLDER/$ASMPREFIX.reads.gm.sam
And can be started simply from the command-line using: ./assemble.sh "FQ file" "PATH TO ASM FOLDER" "PREFIX of output" This finally led to a good assembly after rearranging the middle part which initially was “over-expressed”.
Proposition of new bacteriophage genomes
After having a core genome we want to check how many protein-coding genes we can find on the genome. For this task, again several programs exist. Two of the more common programs are glimmer [7] and genemark [8]. Because the reputation among the target audience is higher for genemark [9] we used this tool for the genome annotation. We ran the tool on the assembled genome in FASTA format generating a gene annotation file (gff3) for the genome highlighting all coding sequences. For easier and more compact usage, we transformed the genome in fasta format with the annotation in gff3 into the embl flat file format. Finally, we can describe the assembled genomes as follows: TABLE
Using the embl flat file format we visualized the phage genomes in a circular genome diagram plot (Figure 5). *-+-+* figures *_*_* Here we can see see several things. For the 3S genome, we can see that at certain position we see a high decrease in the coverage (at 58kbp, 72kbp and 110kbp). At these positions no reads align to the reference genome. For the NES genome we can see a similar behaviour at 330kbp. Additionally we can see two spikes at the ends of the genome. However, we could also see … Finally the FFP genome again has the same problems as the NES genome ends. Some final bliblubb …



Encapsulation
Droplets are Monodisperse



Discussion goes here

Bacteriophages Encapsulated In Alginate Can Withstand Gastric Acid
Discussion goes here
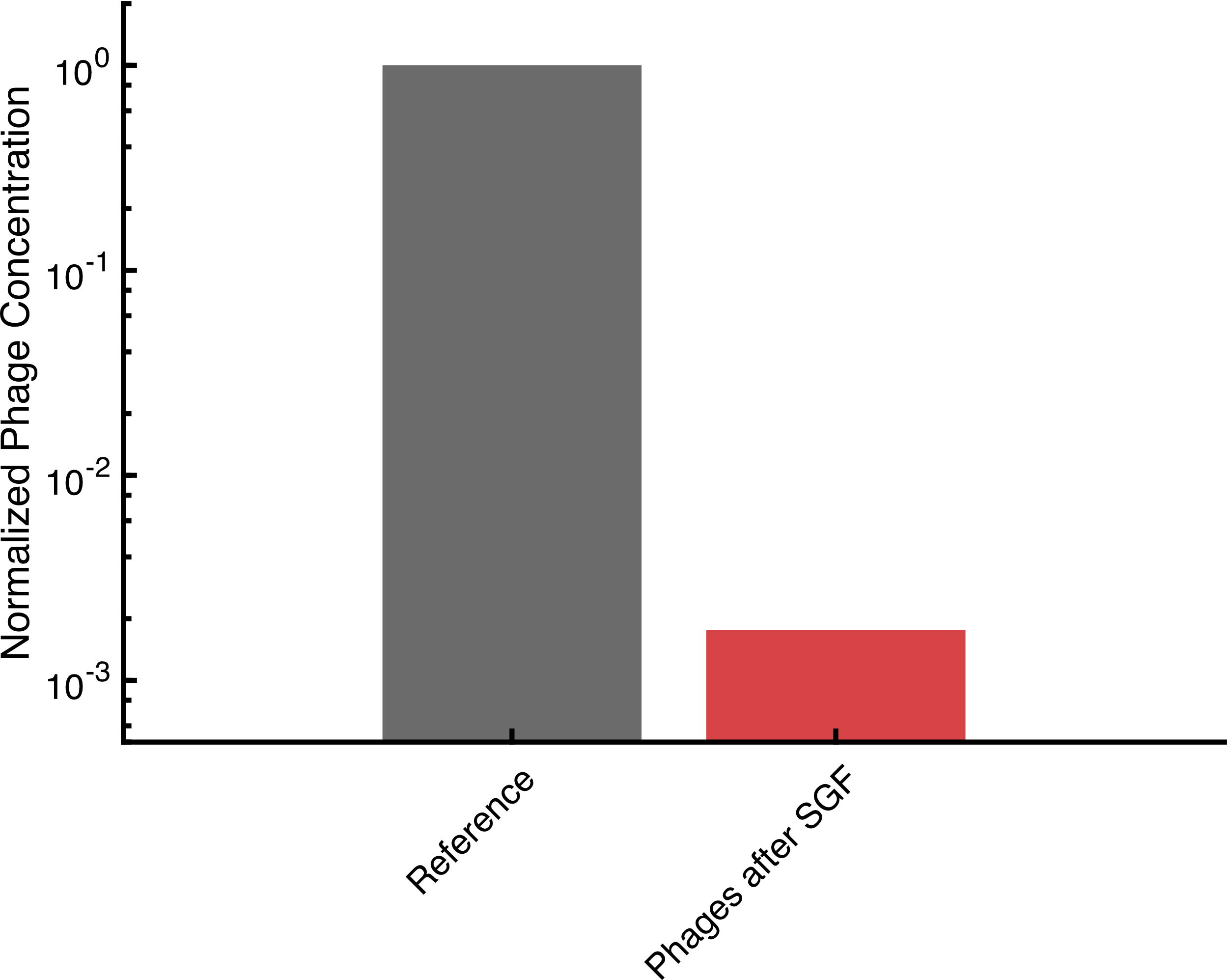
Discussion goes here

Phactory yields phages with toxicity levels that allow oral administration to the patient. However, oral delivery requires protection of the phages from rapid degradation in the acidic gastric juice, while direct intravenous application requires additional purification steps. To overcome these hurdles, we prototyped two 3D-printed fluidic devices that can be assembled for less than $5. For oral application, we built a nozzle to encapsulate the phages in monodisperse calcium-alginate microspheres protecting them in the stomach. The alginate solution, ejected from a dispenser needle with a syringe pump, was sheared off by a parallel stream of air. Our results show that after 1 hour incubation in simulated gastric fluid, active phages are successfully released in simulated intestinal fluid. For intravenous administration, we can purify the bacteriophages from the remaining cell-extract via fractionation in a pressure-driven size-exclusion filter system. Additionally, we built microfluidic hardware for our human practice project OraColi.
- Alginat für lokale pH wert erhöhung im magen und auflösung in chelatoren(darm), da Ca2+ Ionen quervernetzen
- Enkapsulierung durch Co-Flow System. Druckluft schert Tropfen von Spritzennadel ab, Düse == Hardware Quervernetzung 1h in CaCl2 Lsg Waschen Verifikation durch Mikroskopie Fertig für Einsatz (insg. Ca 1.5-2h)
Experimente 1. 3D Modell von SYBR Gold gelabelten Phagen im Droplet durch z-Stack (BF/GFP) 2. Darkfield Bilder von gelabelten Droplets (bild) 3. Droplet Zusammensetzung (Viskosität zweier Alginate) 4. Droplet Größen bei Flussrate und Druck 5. Verhalten im Magen bzw. saurem Millieu (pH: 1) und Pepsin (1h @37°C, 10h @ RT) -> Phagen Degradation (barplot) 6. Verhalten im Darm bzw. pH 7 und Pankreatin (2h @ 37°C) ->Phagen relase über Zeit (plot)



















