Difference between revisions of "Team:Edinburgh UG/Design"
| Line 111: | Line 111: | ||
<div class="col-lg-8 mx-auto"> | <div class="col-lg-8 mx-auto"> | ||
<h1 class="brand-heading">DESIGN</h1> | <h1 class="brand-heading">DESIGN</h1> | ||
| − | |||
<h1 class="brand-heading">Why Do We Need Maxicells?</h1> | <h1 class="brand-heading">Why Do We Need Maxicells?</h1> | ||
<p class="intro-text"></p> | <p class="intro-text"></p> | ||
Revision as of 22:44, 17 October 2018
DESIGN
Why Do We Need Maxicells?
Bacteria and other unicellular prokaryotes may seem somewhat insignificant in the ecosystems we see around us. How can something so small have any influence on the hugely complex multicellular life sharing the environment? We (think we) know all about the bacteria involved in the nitrogen cycle, carbon cycle - bacterial species that we know have a direct affect on the anthropogenic sphere. But what about microbial species that are not directly involved in these processes, should we be concerned about the effects that human activity have on them if it will have no effect on us? The answer is YES.
Recent studies in metagenomics have revealed a complex network of dynamic interactions in microbial communities. Species that were previously thought to be unimportant have been found to impact these processes by providing services to those species performing the key, central reactions. With this knowledge it is clear that any human activity, such as environmental release of reproductive micro-organisms that interfere with natural ecosystem dynamics, can have unknown, possibly long-lasting effects on the biogeochemical cycles which are necessary for life on Earth. It is therefore imperative that any Synthetic Biology venture intended for environmental release should not have any undesired effects on the local ecology.
We considered the possible reasons for why environmental release is so dangerous, and decided that there were two main problems which must be overcome:
- The possibility that our released strain could proliferate uncontrollably
- The risk that genes from our strain could escape by horizontal gene transfer and provide a competitive advantage to native prokaryotes
Both of these outcomes have to potential to alter the balance of ecosystem interaction networks, which could have a myriad of unforeseen consequences.
In order to tackle the first problem, and to a large extent the second, we first looked at minicells: cells generated after abnormal septum formation during replication due to misplacement of the FtsZ ring - leading to the production of small cells that do not possess a chromosome.
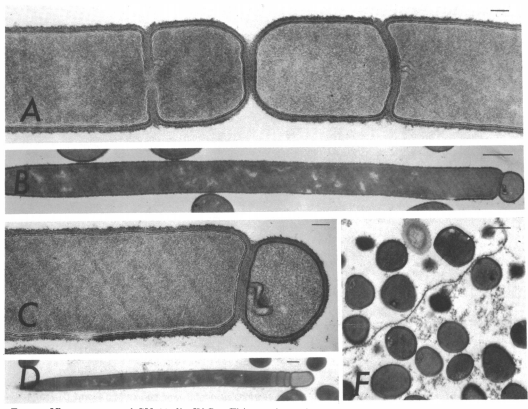
Reeve JN, Mendelson NH, Coyne SI, Hallock LL, Cole RM. Minicells of Bacillus subtilis [Internet]. Vol. 114, JOURNAL OF BACTERIOLOGY. 1973. Available from: http://jb.asm.org/
As they have no chromosome, they are unable to reproduce and proliferate in the environment. Minicells have been shown to retain plasmids, and are therefore presented as a promising novel chassis for Synthetic Biology. It would be possible for minicells to perform any plasmid-encoded function you desire, and would solve the first problem for environmental release. However, minicells presented problems with purification due to their small, inconsistent size. After discussion with experts in the field of biotechnology to try and resolve these issues, it was suggested that we instead try to remove the chromosome by some other means. This suggestion led us to completely change the design of our project; to instead look at maxicells.
Maxicells are bacterial cells which have lost their chromosome, and are therefore unable to reproduce and proliferate in the environment. These maxicells can be used for Synthetic Biology, as they retain any plasmids they possessed prior to chromosome destruction, which we have defined as the “instructor plasmids”. Maxicells would then be as functional and safe as minicells, without the costly, time-consuming purification steps.
While maxicells are unable to reproduce and cannot release any chromosomal genes into the environment, it is nonetheless still possible that advantageous genes from the instructor plasmid could escape by horizontal gene transfer, the most notable of which being the antibiotic resistance marker used for plasmid selection. We have approached this problem in three ways, which we have termed our Triple Lock System:
- The colicin kill switch, which aims to fully degrade any plasmid DNA in the maxicell after a defined, programmable time-frame.
- Our semantic containment system, which ensures that any native prokaryotes will be unable to read and express the instructor plasmid DNA, as it will be written in a different language.
- The alternative selection marker, triclosan, which removes the need for an antibiotic resistance gene to be encoded on the instructor plasmid, thereby preventing the spread of antibiotic resistance.
The design of our project then fell into three overarching sections, which will be detailed below:
- Evaluating and Optimising Maxicell Protocols
- Quantifying the Active Timeframe of Maxicells
- The Triple Lock System
Evaluating and Optimising Maxicell Protocols
We investigated 3 ways in which to produce maxicells in order to present the easiest, most efficient production method for current and future synthetic biologists wishing to use them as a chassis! These three methods were by:
- Ultraviolet light exposure
- Cleaving a palindromic hairpin loop on the chromosome
- Cutting the chromosome with a homing endonuclease
We then attempted to modularise the UV production method by creating knockdowns for DNA repair genes using RNA interference.
UV Production Method
This is the established method of maxicell production, which uses ultraviolet light to damage the DNA within the cell. The exposure to UV light causes pyrimidine dimers to form, which in regular cells would be restored by nucleotide excision repair, however, the maxicell producing strain CSR603 is a knockout for recA - an enzyme involved in DNA repair by homologous recombination - and uvrA - an enzyme involved in nucleotide excision repair. As the cell has no method of repairing the UV-induced damage, the entire chromosome is degraded by exonucleases, leaving an achromosomal maxicell.
We decided to evaluate the efficiency of maxicell production using two Escherichia coli strains: CSR603, the uvrA- recA- strain; and DH5-alpha, a standard recA- lab strain. DH5-alpha was used to test how effective standard, recA+ lab strains are as maxicell producers. After the effectiveness of maxicell production by these strains was investigated, we set out to further increase the modularity of this system by producing a MicC-RecA-UvrA construct to knockdown recA and uvrA using RNA interference. This would allow theoretically any E. coli strain of your choosing to be used as a maxicell producer, and potentially many other bacterial species.
Palindromic Cleavage Method
The next maxicell production method we investigated was suggested to us by experts in Molecular and Cell Biology from the Leach Lab. This uses an E. coli strain developed in their lab, DL 3355, which contains a palindromic site on the chromosome that causes a hairpin loop to form. This hairpin loop may be cleaved by sbcCD exonuclease, causing a double strand break in the DNA. As the strain is recA-, this break cannot be repaired, and the chromosome is degraded by exonucleases.
Homing Endonuclease Method
The last method of maxicell production was also suggested by the Leach Lab, and uses an E. coli strain - DL2524 - that contains an 18 bp recognition site for the homing endonuclease I-SceI, and the I-SceI gene under the control of an arabinose-inducible promoter. When the cells are cultured in 0.5% arabinose, I-SceI is expressed, which then cuts the chromosomal DNA at the recognition site producing a double strand break that leads to complete degradation of the chromosome.
Quantifying Maxicell Timeframe
After we established that we could reliably produce maxicells, we attempted to get a quantitative measurement of how long they could be metabolically active. This would have obvious applications in the field, as you would know the time-frame in which maxicells can be used for your desired process. We took a three-pronged approach to this quantification, beginning by looking at their ATP levels over time, the level of protein retention, and their ability to functionally express a gene at various times after their production.
ATP Quantification
We added the enzymes D-Luciferin and Luciferase to the maxicell and cell samples in order to produce luminescence by hydrolysis of ATP by the enzymes. This luminescence can be directly correlated to the level of ATP in a cell. After calibration with known ATP levels, we used a luminometer to determine the levels of ATP in our maxicells over a 72 hour period at 4 °C, 25 °C and 37 °C. This would show us how long maxicells could be stored in the refrigerator and how long they could be active for at room temperature and human body temperature.
Protein Retention
We next looked at the protein levels within maxicells at various time points and concentrations. We figured that the half-life of proteins would be much longer than in non-maxicells, as the half-life would no longer be intrinsically tied to the rate of division; in normal cells the level of protein halves at every cell-division. The protein levels within maxicells would therefore be solely dependent on protein export and degradation rate. These values were unknown in maxicells, as they have an altered metabolism, so we sought to determine the overall protein levels using SDS-PAGE and compare them at different time-points. As with ATP quantification, this was performed at 4 °C, 25 °C and 37 °C.
Functional Gene Expression
The above characterisations give us some idea of how long maxicells can operate, but we considered that there may be another, unknown factor that affects their functionality. As a definitive test to determine their functional lifespan, we decided to insert a construct into our maxicell instructor plasmid to see how well it can express a gene after a given time. We settled on two constructs:
- ars-mCherry - the fluorescent protein mCherry under control of an arsenite inducible promoter
- araC-mCherry - mCherry under an arabinose promoter
The added benefit of using these parts was to test how effectively our maxicells can function as a biosensor; in these cases for arsenic and arabinose. This would be a characterisation of how well maxicells actually work in the field, for one of their potential applications. We planned to transform the maxicell producing E. coli strain CSR603 with these constructs and then induce mCherry expression with arsenic or arabinose after 18 hours, 19 hours and 24 hours. This would give us a better idea of how long they can survive and still have the potential for gene expression.
The Triple Lock System
Colicin Kill Switch
As the first ‘lock’ in our triple lock system, the function of the colicin kill switch is to prevent the instructor plasmid from being released from the maxicell into the environment in the first place. This 2-part system was inspired by Darmstadt 2016 iGEM team, but altered and optimised for use in maxicells. The kill switch works by the action of colicin E2, a nicking endonuclease which cuts single and double stranded DNA at random sites. The counterpart to E2 is Imm2; an immunity protein which binds to E2 and inhibits its endonuclease activity. Imm2 binds in a 1:1 stoichiometric ratio, so if the level of Imm2 in the cell should fall below the level of E2, then E2 is free to function. We hypothesised that by fine-tuning of the expression of these genes, we could cause the Imm2 concentration to drop below that of E2 at a defined time. This time point would be the point at which the instructor plasmid would start to be degraded. We therefore made some alteration to Darmstadt's design to make it applicable for the release of maxicells.
The first alteration was to the Immunity protein construct (Imm2). We have inserted I-Sce1 sites flanking the imm2 gene in the plasmid. This makes the system compatible with the ‘Homing Endonuclease’ (I-SceI) method of maxicell production. When I-SceI expression is induced to digest the chromosome, the imm2 gene will also be cut out and therefore is no longer expressed. Imm2 protein levels will fall over time until Colicin-E2 protein is no longer bound by Imm2 and is free to digest the plasmid.
Following environmental release, conditions for the maxicell will be uncontrolled and variable. Therefore, the longer the maxicell is in the environment, the higher the probability of a chance maxicell lysis event, and release of the plasmid into the surroundings. The plasmid may then be picked up by other prokaryotes in the environment. It is important that the instructor plasmid is degraded before this occurs. However, we do not want to inhibit maxicell function (whatever that may be) by destroying the instructor plasmid too early. Ideally, plasmid degradation should occur immediately prior the to the end of the active metabolic lifetime of the maxicell. Timing of plasmid destruction by Colicin should be finely tuned meaning selection of promoters and ribosome binding sites for E2 and Imm2 expression is crucial in order for degradation to occur at the desired point in time.
We have used rational design in order to select the promoters and ribosome binding sites necessary for triggering plasmid degradation at the point we estimate our maxicells will no longer be metabolically active (see 'Quantifying Maxicell Timeframe' above). We have designed a model that predicts the degradation time point for every possible promoter-RBS combination for Colicin and Imm2 using the Anderson promoters and ribosome binding sites on the iGEM registry. From this model we selected promoter BBa_J23105 for Imm2, and promoter BBa_J23106 for Colicin E2. Both parts had RBS BBa_J61107 and Terminator BBa_B0010

Semantic Containment
The second lock in our triple lock system ensures that even if the first lock fails, an environmental cell would not be able to express and utilise a gene obtained from our maxicells. The basis for this came from the Paris-Bettencourt 2012 project but we have taken it a step further. This involved rewriting the DNA of our instructor plasmid so that it cannot be read by a foreign organism. Essentially we're giving our plasmid a different language.
The basis of semantic containment lies in the mutation of the supD gene - a tRNA amber suppressor - which causes amber STOP codons to be read as a serine codon instead. Our sequence may be re-coded to change serine codons to amber STOP codons, and as long as it has the supD mutation the genes will be translated correctly. However, if a foreign organism were to take up the DNA, it would read the amber codons as a STOP, producing a non-functional, truncated protein. This was achieved by Paris-Bettencourt using one and two serine codon replacements with amber STOP codons in the kanamycin resistance gene, which they designated P1003* and P1003**. This allows easy quantification of the effective expression of recoded genes by non-supD mutants simply by testing growth rates on kanamycin. Paris-Bettencourt's results showed that non-supD mutants with the recoded P1003* and P1003** genes showed limited growth on kanamycin, suggesting that the recoding was successful to a certain extent. We decided, however, that these results did not show a high enough level of security, and so we planned to go a step further.
Before designing parts for this, we again approached the problem by utilising rational design. We modelled the probability of a translational read-through with increasing numbers of amber stop codons present in a coding sequence. With 2 amber codons, 1 in every 10,000 translation attempts will be able to generate a read through and express the recoded gene. When aiming for complete safety and containment, this is not good enough… We therefore designed 2 new parts building on Paris-Bettencourt’s: P1003 5* [BBa_K2725012] and P1003 10* [BBa_K2725013] . These have 5 and 10 serine codons from the P1003 coding sequence substituted with amber stop codons. With 5 amber codons present in a coding sequence, probability of a read through and subsequent expression is reduced to 1 in every 1010 translation attempts, and with 10 amber codons this is reduced further to 1 in 1018 translation attempts. For more information on our probability model, see our semantic containment modelling page.
Addition of more amber codons however, also presents a problem for expression of the recoded gene in our chassis. In order to solve this we used the same probability model in order to maximise read through in our chassis. We took the supD + terminator sequence from Paris-Bettencourt's part, BBa_K914000 and assembled this downstream of a higher strength constitutive Anderson promoter. Higher levels of amber suppressor supD increases the likelihood of the tRNA interacting with an amber codon instead of Release Factor 1 that would release the nascent polypeptide from the ribosome and halt translation. We designed 3 of these parts with varying strength anderson promoters in order to test for proof of concept:
- J23102-supD - [BBa_K2725014] (strength 0.82)
- J23103-supD - [BBa_K2725015] (strength 0.01)
- J23108-supD - [BBa_K2725016] (strength 0.51)
Alternative Selection
The third lock in our triple lock system ensures that even if an environmental cell takes up an instructor plasmid, and can read and express the genes present, that those genes confer no competitive advantage to the environmental cell. This is done by replacing the antibiotic resistance gene in the standard iGEM backbone with a gene that gives resistance to the biocide, triclosan. Triclosan is a chemical that has previously been used in health and beauty products such as toothpaste, but today it is very rarely used. Critically, it is not used in medicine and healthcare, therefore if a pathogen became triclosan resistant this would not interfere in our ability to treat such an infection. The triclosan resistance gene, FabV, offers no cross resistance to any commonly used antibiotics. It is also cheaper than conventional antibiotics as lower concentrations need to be used for selection: <1p per litre compared to 56p per litre for ampicillin (prices from Alfa Aesar).
Triclosan is a biocide which acts upon FabI protein in E. coli, an enoyl ACP-reductase involved in fatty acid biosynthesis. We planned to investigate the effectiveness of resistance by two mechanisms. The first is by overexpression of fabI, which gives triclosan resistance as there is so much of the target that no matter how much FabI is broken down, there is still enough for the system to function. The second method of resistance is by heterologous expression of fabV – an enoyl ACP-reductase found in Vibrio species. Research has shown that FabV confers greater resistance than FabI, as it is refractory to inhibition by triclosan.
Contact EdiGEM18
Feel free to leave us a comment on social media!