Difference between revisions of "Team:Bielefeld-CeBiTec/Software"
| Line 186: | Line 186: | ||
With all defined formulas \((2)\),\((3)\) and \((4)\), formula \((1)\) can now be calculated as follows: | With all defined formulas \((2)\),\((3)\) and \((4)\), formula \((1)\) can now be calculated as follows: | ||
| − | $$P(eff|X) = \frac{P^{eff} P(X|eff)}{P^{eff} P(X|eff)+P^{inf} P(X|inf)} \\= \frac{P^{eff} \prod_{i=1}^{19} q_{x_i^n}^{eff}}{P^{eff} \prod_{i=1}^{19} q_{x_i^n}^{eff}+P^{inf} \prod_{i=1}^{19} q_{x_i^n}^{inf}} $$ | + | $$P(eff|X) = \frac{P^{eff} P(X|eff)}{P^{eff} P(X|eff)+P^{inf} P(X|inf)} \\ \\= \frac{P^{eff} \prod_{i=1}^{19} q_{x_i^n}^{eff}}{P^{eff} \prod_{i=1}^{19} q_{x_i^n}^{eff}+P^{inf} \prod_{i=1}^{19} q_{x_i^n}^{inf}} $$ |
</article> | </article> | ||
Revision as of 09:13, 17 October 2018


siRCon - A siRNA Constructor
siRNAS short introduction
Choosing appropriate design methods

Rational siRNA design
| Rule | Score |
|---|---|
| 30%-52% G/C content | +1 |
| At least 3 'A/U' bases at positions 15-19 | +1 (for each 'A/U' base) |
| Absence of internal repeats (\(T_m \lt 20\)) | +1 |
| An 'A' base at position 3 | +1 |
| An 'A' base at position 19 | +1 |
| An 'U' base at position 19 | +1 |
| A base other than 'G' or 'C' at 19 | -1 |
| A base other than 'G' at position 13 | -1 |
Ui-Tei rule
- An ‘A’ or ‘T’ at position 19
- A ‘G’ or ‘C’ at position 1
- At least five ‘U’ or ‘A’ residues from positions 13 to 19
- No ‘GC’ stretch more than 9nt long
Calculating silencing probability
siRNA overhangs and scaffolds


Check siRNA
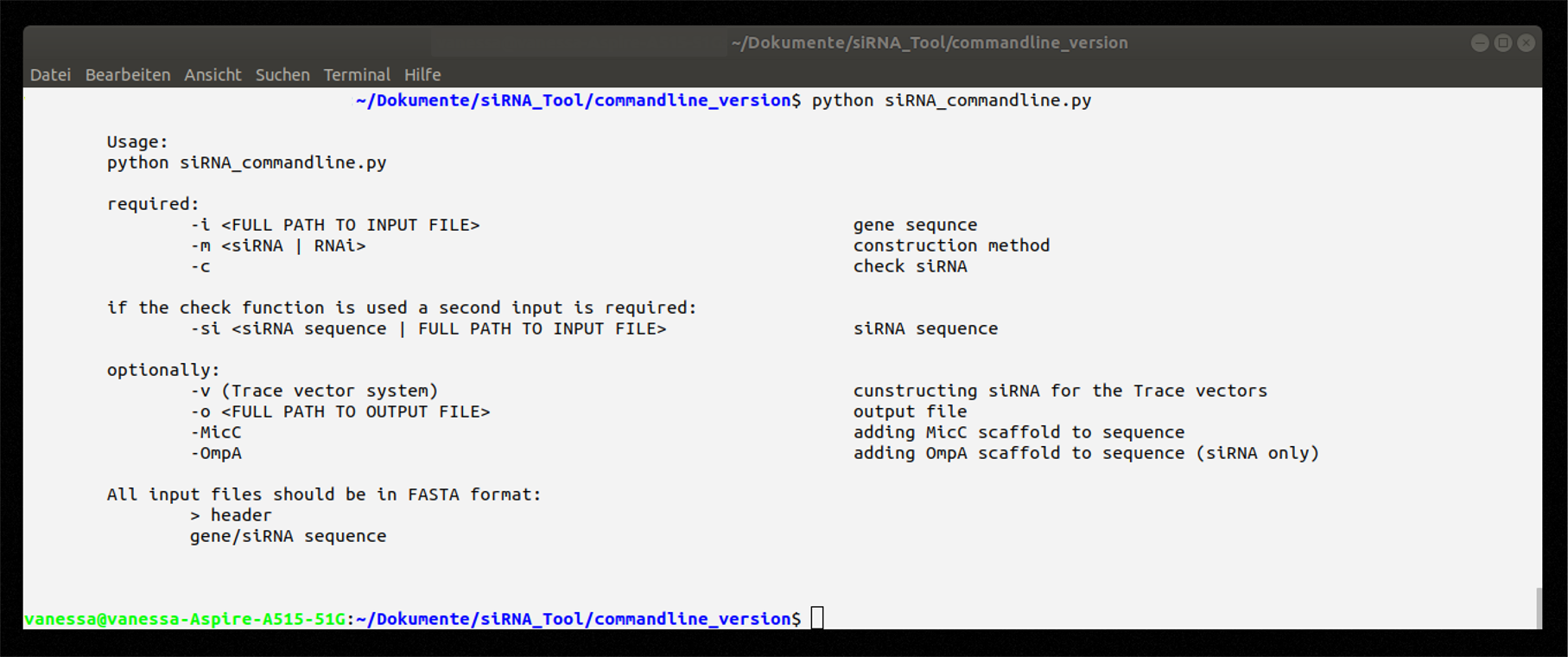
Command line application
Graphical Interface usage

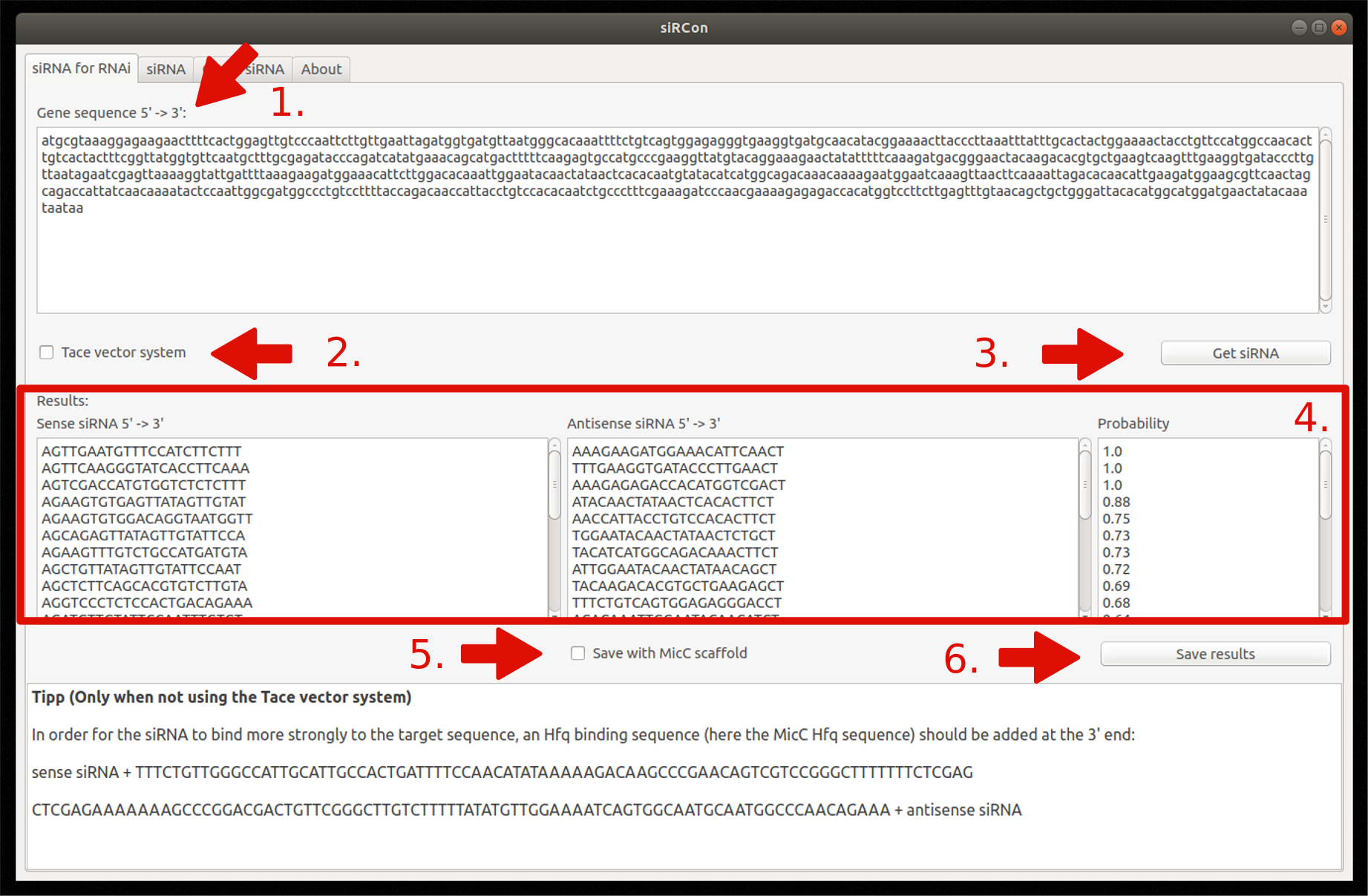
1. siRNA for RNAi
- Insert gene sequence
- Choose Tace vector system (optionally)
- Constructions of siRNAs
- View resulting siRNAs (sense and antisense sequence) and their corresponding probability
- Decide if siRNAs should be saved with MicC scaffold (only if Tace is not used)
- Save results as FASTA file
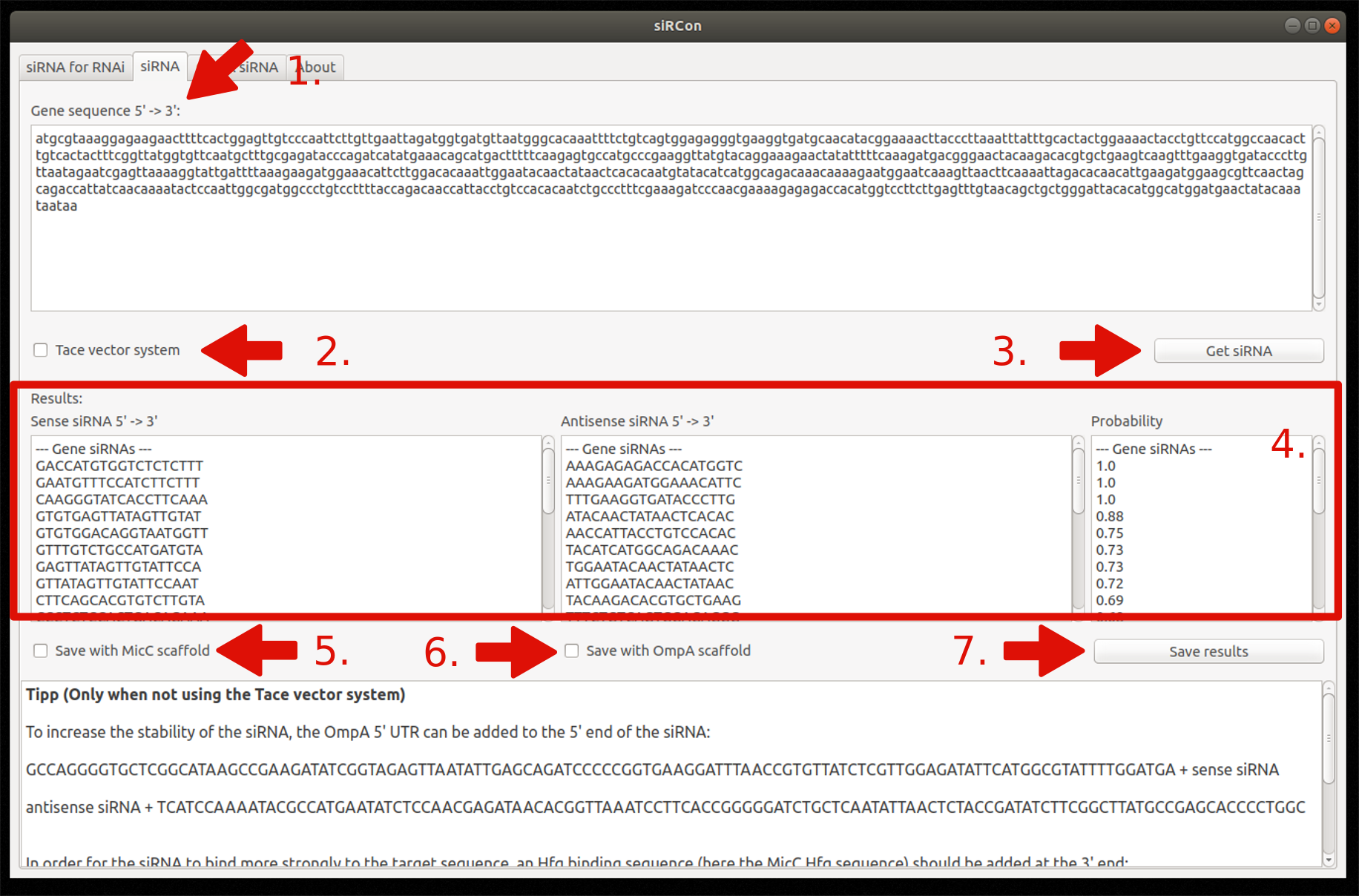
2. siRNA for silencing
- Insert gene sequence
- Choose Tace vector system (optionally)
- Constructions of siRNAs
- View resulting siRNAs (sense and antisense sequence) and their corresponding probability
- Decide if siRNAs should be saved with MicC scaffold (only if Tace is not used)
- Decide if siRNAs should be saved with OmpA scaffold (only if Tace is not used)
- Save results as FASTA file
3. Check siRNA
- Insert gene sequence
- Insert siRNA sequences
- Choose method the siRNA was constructed for (siRNA for RNAi or siRNA for silencing)
- Choose if siRNA was constructed for Tace (optionally)
- Validation of entered siRNA for given target gene sequences
- View results
- Save results (optionally)
