Difference between revisions of "Team:Vilnius-Lithuania-OG/Design"
| Line 242: | Line 242: | ||
<p>After the regulatory parts are added the fragments must then be <strong>circularized (3)</strong>. DNA fragments are circularized for two main reasons:</p> | <p>After the regulatory parts are added the fragments must then be <strong>circularized (3)</strong>. DNA fragments are circularized for two main reasons:</p> | ||
| − | <p class="listin">1. First of all, in order to amplify the DNA inside microfluidic droplets an <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="Isothermal amplification reactions covers multiple methods which utilize the amplification reaction at a single-temperature, usually are extremely fast and do not require thermocyclers">isothermal amplification reaction</a> must be used. That is because droplets become highly unstable and can break easily when temperature is quickly | + | <p class="listin">1. First of all, in order to amplify the DNA inside microfluidic droplets an <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="Isothermal amplification reactions covers multiple methods which utilize the amplification reaction at a single-temperature, usually are extremely fast and do not require thermocyclers">isothermal amplification reaction</a> must be used. That is because droplets become highly unstable and can break easily when the temperature is quickly altered - just like during the popular polymerase chain reaction (PCR). If droplets would break during the initial stages of amplification, all of the information from different droplets would get mixed in the tube and the knowledge about which activity belongs to which sequence would be lost, as it was not yet recorded by the means of amplification (before the amplification, the activity measurement information is in form of modified substrate nucleotides).<br /> That is why CAT-Seq uses a multiple displacement amplification (MDA), which is a non-thermocycling (isothermal) based DNA amplification technique. MDA was chosen because of its ability to rapidly amplify single templates of DNA into long double-stranded molecules which contain multiple repeats of the starting template. That said, one of the core requirements of MDA reaction is that starting DNA template <strong>must be circular</strong>.</p> |
<p class="listin">2. Secondly, as seen in the later steps of CAT-Seq, in order to efficiently express a catalytic biomolecule in droplets, <strong>Rolling Cycle Transcription</strong> (described in catalytic biomolecule production section) is used, which immensely increases mRNA synthesis yield from a single DNA template. Such technique also <strong>requires the DNA to be circular</strong>, as an RNA polymerase travels around it in circles producing mRNA.</p> | <p class="listin">2. Secondly, as seen in the later steps of CAT-Seq, in order to efficiently express a catalytic biomolecule in droplets, <strong>Rolling Cycle Transcription</strong> (described in catalytic biomolecule production section) is used, which immensely increases mRNA synthesis yield from a single DNA template. Such technique also <strong>requires the DNA to be circular</strong>, as an RNA polymerase travels around it in circles producing mRNA.</p> | ||
| Line 253: | Line 253: | ||
<p><strong>Obtaining the first catalytic biomolecule</strong></p> | <p><strong>Obtaining the first catalytic biomolecule</strong></p> | ||
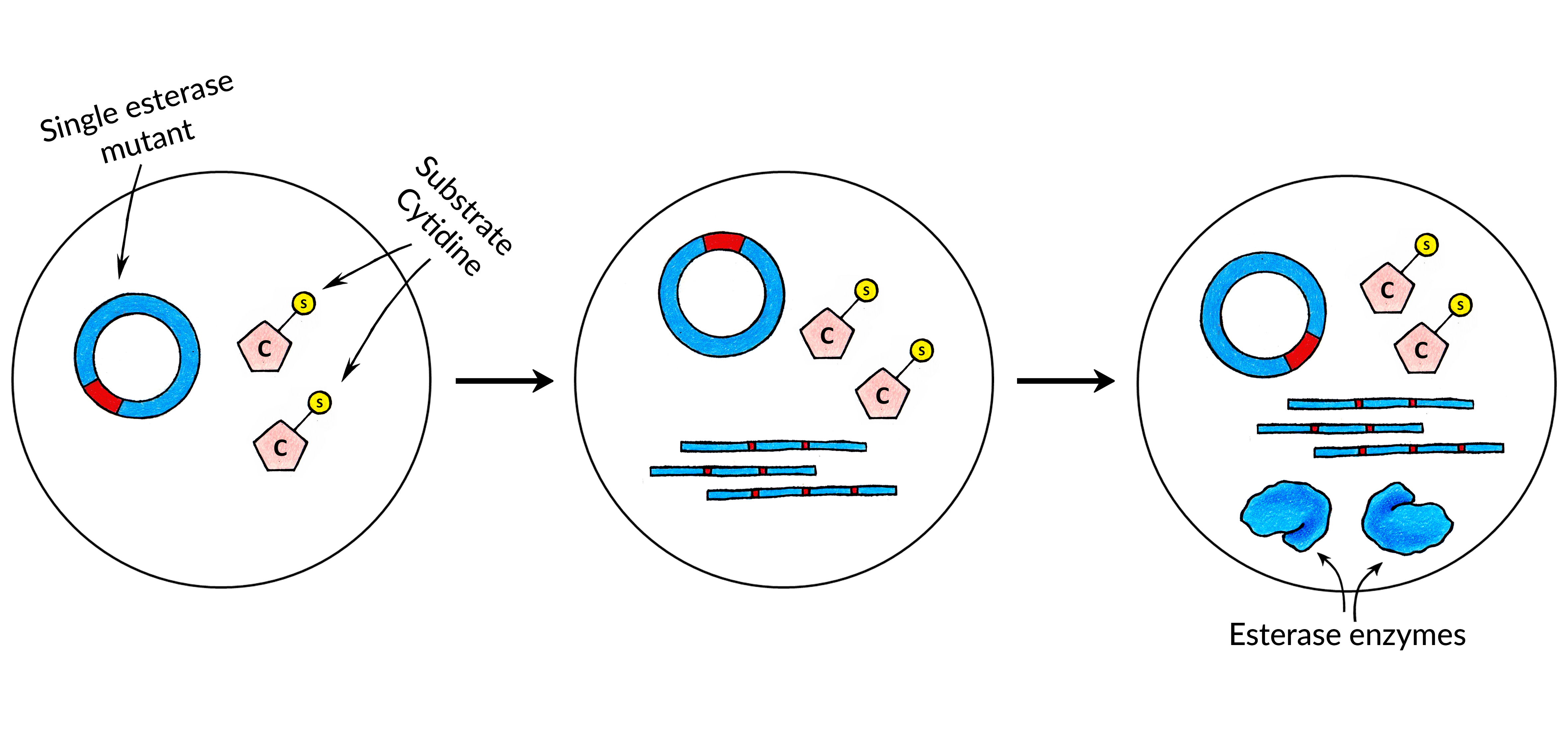
<p>In order to find a correct combination of Substrate Nucleotide and the Catalytic Biomolecule that can catalyze the removal of the substrate, we have decided to look for a hydrolase. The is because a direct removal of the substrate from nucleotide seemed like the most timewise efficient choice.</p> | <p>In order to find a correct combination of Substrate Nucleotide and the Catalytic Biomolecule that can catalyze the removal of the substrate, we have decided to look for a hydrolase. The is because a direct removal of the substrate from nucleotide seemed like the most timewise efficient choice.</p> | ||
| − | <p> First, we synthesized a cytidine which had a benzoyl substrate attached to it: <strong>N4-benzoyl-2'-deoxycytidine triphosphate</strong>. Instructions | + | <p> First, we synthesized a cytidine which had a benzoyl substrate attached to it: <strong>N4-benzoyl-2'-deoxycytidine triphosphate</strong>. Instructions on how such substrate nucleotide can be synthesized can be found here.</p> |
<p>Then, an <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="An esterase is a hydrolase enzyme that splits esters into an acid and an alcohol in a chemical reaction with water called hydrolysis">esterase</a> library was screened manually using microwell plates in order to find the hydrolase enzyme which can catalyse the removal of the substrate. Once such enzyme was found and its sequence was determined. We have then used this pair of <strong>esterase enzyme</strong> and <strong>substrate cytidine</strong> to build the first version of CAT-Seq.</p> | <p>Then, an <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="An esterase is a hydrolase enzyme that splits esters into an acid and an alcohol in a chemical reaction with water called hydrolysis">esterase</a> library was screened manually using microwell plates in order to find the hydrolase enzyme which can catalyse the removal of the substrate. Once such enzyme was found and its sequence was determined. We have then used this pair of <strong>esterase enzyme</strong> and <strong>substrate cytidine</strong> to build the first version of CAT-Seq.</p> | ||
| Line 260: | Line 260: | ||
| − | <p>Next, we have performed an in-silico modelling of our Esterase enzyme. The goal of the in-silico analysis was to create the Esterase mutants which would have | + | <p>Next, we have performed an in-silico modelling of our Esterase enzyme. The goal of the in-silico analysis was to create the Esterase mutants which would have different catalytic activities. Once such mutants were constructed, their relative catalytic activities were measured again using the same standard methods we have used to find the initial esterase enzyme. This was done in order to construct a small Esterase mutant library which would have precisely-defined catalytic activities by already established low-throughput measuring methods. Then, with CAT-Seq, we can also measure the activities of those mutants in high-throughput, which can then be <strong>compared to our previous measurements</strong> to confirm that CAT-Seq system is working as intended and can precisely discriminate different mutant activities.</p> |
| − | <p> After acquiring the esterase enzyme and its mutants, <strong>regulatory sequences </strong>- RNA polymerase promoter and ribosome binding site - were added to each of the library | + | <p> After acquiring the esterase enzyme and its mutants, <strong>regulatory sequences </strong>- RNA polymerase promoter and ribosome binding site - were added to each of the library members. Then, the fragments were <strong>circularized </strong>for the MDA and RCT reactions.</p> |
<p><strong>After acquiring the initial esterase for building CAT-Seq, why were the mutations added manually and not randomly?</strong></p> | <p><strong>After acquiring the initial esterase for building CAT-Seq, why were the mutations added manually and not randomly?</strong></p> | ||
<p>The goal of the in-silico modelling of the esterase was to determine mutants that would have different catalytic activities. If we had a randomized library of esterase mutants, it would have been difficult to precisely tune CAT-Seq parts as we would not be sure what output of the system we should expect. In contrast, having mutants with precisely measured catalytic activities beforehand using well-established and precise low-throughput methods enables us to recognise how well our system is performing. </p> | <p>The goal of the in-silico modelling of the esterase was to determine mutants that would have different catalytic activities. If we had a randomized library of esterase mutants, it would have been difficult to precisely tune CAT-Seq parts as we would not be sure what output of the system we should expect. In contrast, having mutants with precisely measured catalytic activities beforehand using well-established and precise low-throughput methods enables us to recognise how well our system is performing. </p> | ||
| Line 307: | Line 307: | ||
<img class="image_design" style="padding-bottom: 4%;; width: 30%; margin: auto; display: block;" src="https://static.igem.org/mediawiki/2018/3/37/T--Vilnius-Lithuania-OG--poisson_formula.png"> | <img class="image_design" style="padding-bottom: 4%;; width: 30%; margin: auto; display: block;" src="https://static.igem.org/mediawiki/2018/3/37/T--Vilnius-Lithuania-OG--poisson_formula.png"> | ||
| − | <p>P(X = k) is the probability for single droplet to house k DNA molecules. λ shows the average number of DNA molecules per droplet. For example, if you want to have a single template per droplets, you might choose λ to be 1. The Poisson distribution predicts, that 36.7% of droplets will have one molecule. However, it also predicts that 18.4% of droplets will house 2 molecules and 6.1% - 3 molecules. Following this notation, In order to avoid artificial data, caused by multiple DNA templates in one droplet, the concentration of DNA template was kept at 1 per 10 droplets (λ = 0.1). As seen from the graph, 90.4% of droplets will be empty and 9% will have 1 template. On the other hand, 0.4%. of droplets will house 2 DNA molecules, meaning that droplets with 2 DNA templates are generated rarely</p> | + | <p>P(X = k) is the probability for the single droplet to house k DNA molecules. λ shows the average number of DNA molecules per droplet. For example, if you want to have a single template per droplets, you might choose λ to be 1. The Poisson distribution predicts, that 36.7% of droplets will have one molecule. However, it also predicts that 18.4% of droplets will house 2 molecules and 6.1% - 3 molecules. Following this notation, In order to avoid artificial data, caused by multiple DNA templates in one droplet, the concentration of the DNA template was kept at 1 per 10 droplets (λ = 0.1). As seen from the graph, 90.4% of droplets will be empty and 9% will have 1 template. On the other hand, 0.4%. of droplets will house 2 DNA molecules, meaning that droplets with 2 DNA templates are generated rarely</p> |
<img class="image_design" style="margin-bottom: 3%; width: 100%; margin: auto; display: block;" src="https://static.igem.org/mediawiki/2018/8/89/T--Vilnius-Lithuania-OG--graph.png"> | <img class="image_design" style="margin-bottom: 3%; width: 100%; margin: auto; display: block;" src="https://static.igem.org/mediawiki/2018/8/89/T--Vilnius-Lithuania-OG--graph.png"> | ||
| Line 330: | Line 330: | ||
<img class="image_design" style="margin-bottom: 3%;display: block;" src="https://static.igem.org/mediawiki/2018/9/9f/T--Vilnius-Lithuania-OG--no_protein.jpeg"> | <img class="image_design" style="margin-bottom: 3%;display: block;" src="https://static.igem.org/mediawiki/2018/9/9f/T--Vilnius-Lithuania-OG--no_protein.jpeg"> | ||
| − | <p>After doing in-depth literature research to try | + | <p>After doing in-depth literature research to try to solve this problem, we have found out that around 1990s people immensely increased short microRNA synthesis yields by circularizing DNA templates. As RNA polymerase goes around the DNA in circles it produces long strands of RNA molecules. Within those long molecules were the repeating small units of microRNA. They coined this method <strong>Rolling Circle Transcription</strong> (RCT). </p> |
| Line 400: | Line 400: | ||
| − | <p>Another important thing to consider is creating a reference point which would help to precisely determine the | + | <p>Another important thing to consider is creating a reference point which would help to precisely determine the activities of different catalytic biomolecules. For CAT-Seq, such reference point is nucleotides with reference modification. This modification can be any small modification which still allows a DNA polymerase to incorporate it during the amplification. We call them the <strong>Reference Nucleotides. </strong></p> |
| − | <p>For example, If we would try to amplify DNA template in each droplet that contains different amounts of nucleotides that can be incorporated, we would get a sequencing <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="Bias is disproportionate weight in | + | <p>For example, If we would try to amplify DNA template in each droplet that contains different amounts of nucleotides that can be incorporated, we would get a sequencing <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="Bias is disproportionate weight in favour of or against one thing compared with another">bias</a> that would be more favourable for biomolecules that are highly active.</p> |
| Line 409: | Line 409: | ||
| − | <p>That is because biomolecules with higher activity would produce more nucleotides for polymerase and the amplified DNA amount would be much bigger | + | <p>That is because biomolecules with higher activity would produce more nucleotides for polymerase and the amplified DNA amount would be much bigger than compared to lower activity mutants which would produce less. Reference nucleotides can easily remove this bias - after merging the Substrate Droplet with Amplification droplets that contain Reference Nucleotides, they allow the synthesis of a similar amount of amplified DNA in each droplet.</p> |
| Line 430: | Line 430: | ||
<p>We now have a population of droplets, wherein each droplet contains a <strong>specific amount of Product Nucleotides</strong> or 2'-deoxycytidine triphosphates.</p> | <p>We now have a population of droplets, wherein each droplet contains a <strong>specific amount of Product Nucleotides</strong> or 2'-deoxycytidine triphosphates.</p> | ||
| − | <p>As already mentioned earlier, we have chosen to use MDA reaction in order to amplify the DNA isothermally. Isothermal amplification allows us to amplify the DNA without disrupting the stability of the droplets. Also, for the Reference Nucleotides we have decided to use Methylated Cytidines (2'-deoxy-5-methylcytidine 5'-triphosphates). These Reference Nucleotides can be readily incorporated by multiple polymerases, including Phi29 which we use for our MDA reaction.</p> | + | <p>As already mentioned earlier, we have chosen to use MDA reaction in order to amplify the DNA isothermally. Isothermal amplification allows us to amplify the DNA without disrupting the stability of the droplets. Also, for the Reference Nucleotides, we have decided to use Methylated Cytidines (2'-deoxy-5-methylcytidine 5'-triphosphates). These Reference Nucleotides can be readily incorporated by multiple polymerases, including Phi29 which we use for our MDA reaction.</p> |
<p>Next, we need to merge our Substrate Droplets with the Amplification Droplets containing the MDA reaction mix. Droplet merging is not an easy task to achieve, as efficient merging requires precise tuning of multiple parameters, such as 4 different droplet and oil flow rates and the strength of the electrical field that helps the droplets to merge. Searching for these parameters experimentally can often be like searching for a needle in a haystack. For this reason, before starting to do droplet merging experiments in the laboratory, we have written a thorough mathematical model which describes the droplet merging in detail and has the ability to predict the correct experimental parameters. We have successfully determined those <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="Water and oil phase flow rates, different droplet volume ratios, the strength of the electrical field and other">parameters</a> and showed experimentally, that they were indeed well-performing conditions for the droplet merging.</p> | <p>Next, we need to merge our Substrate Droplets with the Amplification Droplets containing the MDA reaction mix. Droplet merging is not an easy task to achieve, as efficient merging requires precise tuning of multiple parameters, such as 4 different droplet and oil flow rates and the strength of the electrical field that helps the droplets to merge. Searching for these parameters experimentally can often be like searching for a needle in a haystack. For this reason, before starting to do droplet merging experiments in the laboratory, we have written a thorough mathematical model which describes the droplet merging in detail and has the ability to predict the correct experimental parameters. We have successfully determined those <a href="javascript:;" data-toggle="tooltip" title="" data-original-title="Water and oil phase flow rates, different droplet volume ratios, the strength of the electrical field and other">parameters</a> and showed experimentally, that they were indeed well-performing conditions for the droplet merging.</p> | ||
<p>Substrate Droplets could then be merged with Amplification Droplets containing MDA mix and Reference Nucleotides.</p> | <p>Substrate Droplets could then be merged with Amplification Droplets containing MDA mix and Reference Nucleotides.</p> | ||
| − | <p>Each merged droplet now contains a specific ratio of Product Cytidines to Methylated Cytidines. Such information can then | + | <p>Each merged droplet now contains a specific ratio of Product Cytidines to Methylated Cytidines. Such information can then be recorded by initiating MDA reaction. After the reaction, the amplified DNA molecules now have the specific ratio of nucleotides, wherein the ratio of those nucleotides depend on the specific esterase mutant activity.</p> |
| Line 449: | Line 449: | ||
<p>The last step of the CAT-Seq is the retrieval of recorded activity information from the amplified DNA. In theory, this may be achieved by any sequencing method that can incorporate a DNA modification recognition step. Yet, CAT-Seq uses <strong>Nanopore Sequencing</strong> for several reasons:</p> | <p>The last step of the CAT-Seq is the retrieval of recorded activity information from the amplified DNA. In theory, this may be achieved by any sequencing method that can incorporate a DNA modification recognition step. Yet, CAT-Seq uses <strong>Nanopore Sequencing</strong> for several reasons:</p> | ||
| − | <p class="listin">1. Nanopore sequencing allows to sequence<strong> long molecules</strong>, which eliminates the need of biomolecule fragmentation. This is an extremely important characteristic us | + | <p class="listin">1. Nanopore sequencing allows to sequence<strong> long molecules</strong>, which eliminates the need of biomolecule fragmentation. This is an extremely important characteristic to us because when working with large libraries of mutants in single droplets, fragmenting amplified DNA in those droplets means that all of those fragments in droplets would need to be barcoded. While in theory, this is a possible thing to do, it would require an extra merging and amplification step which would immensely decrease the accuracy of the activity determination. That is because in general, every step of the biotechnological method never is one hundred percent efficient. In turn, the lower amount of steps your system needs to take in order to produce results - the more accurate it will be. That is why reducing the number of steps in CAT-Seq by <strong>omitting fragmentation and barcoding</strong> was extremely important for us.</p> |
<p class="listin">2. Secondly, nanopore sequencing can detect and discriminate different modified nucleotides with ease, because nanopore can detect slight changes in nucleotide structure and assign a different value to that signal.</p> | <p class="listin">2. Secondly, nanopore sequencing can detect and discriminate different modified nucleotides with ease, because nanopore can detect slight changes in nucleotide structure and assign a different value to that signal.</p> | ||
Revision as of 21:35, 17 October 2018