In our project we introduce RNA interference (RNAi) and silencing with small interfering (si)RNAs as an alternative to CRISPR/Cas. To use siRNA as silencing agents for the gene-of -interest we propose a two-step design process. At first potential siRNAs for prokaryotic organisms must be designed. In the second step the silencing effect of these siRNAs can be validated by our siRNA vector system <a href"https://2018.igem.org/Team:Bielefeld-CeBiTec/siRNA">Tace.</a> To facilitate the initial siRNA design step, we developed a siRNA construction tool, which can find possible siRNAs for a given gene sequence and calculate their gene silencing probability. It consists of the three modules siRNAs for RNAi, siRNA, and check siRNA. Obtained siRNAs are perfectly compatible with our siRNA vector system. To the best of our knowledge, this is the first tool dedicated to predicting customized siRNA for the application in prokaryotes. This Python tool comes in two versions: a command line application and an easy-to-use graphical interface.
+
In our project we introduce RNA interference (RNAi) and transcription repression with small interfering (si)RNAs as an alternative to CRISPR/Cas. To use siRNA as silencing agents for the gene-of -interest we propose a two-step design process. At first potential siRNAs for prokaryotic organisms must be designed. In the second step the silencing effect of these siRNAs can be validated by our siRNA vector system <a href"https://2018.igem.org/Team:Bielefeld-CeBiTec/siRNA">Tace.</a> To facilitate the initial siRNA design step, we developed a siRNA construction tool, which can find possible siRNAs for a given gene sequence and calculate their gene silencing probability. It consists of the three modules siRNAs for RNAi, siRNA, and check siRNA. Obtained siRNAs are perfectly compatible with our siRNA vector system. To the best of our knowledge, this is the first tool dedicated to predicting customized siRNA for the application in prokaryotes. This Python tool comes in two versions: a command line application and an easy-to-use graphical interface.
</article>
</article>
Line 103:
Line 103:
<article>
<article>
−
siRNAs are small single- or double-stranded RNAs with an average length of 21-25 nucleotides. They are non-coding RNAs which can bind a specific complementary coding mRNA and silence its function. During eukaryotic RNAi siRNAs are loaded to Argonaute proteins, which carry out the repression, either by blocking mRNA translation or by degrading the mRNA (Siomi and Siomi, 2009). More about the siRNAs and the mechanisms are found <a href="https://2018.igem.org/Team:Bielefeld-CeBiTec/siRNA">here.</a>
+
siRNAs are small single- or double-stranded RNAs with an average length of 21-25 nucleotides. They are non-coding RNAs which can bind a specific complementary coding mRNA and silence its function. During eukaryotic RNAi siRNAs are loaded to Argonaute proteins, which carry out the repression, either by blocking mRNA translation or by degrading the mRNA (Siomi and Siomi, 2009). More detailed information on both possible siRNAs mechanisms are found <a href="https://2018.igem.org/Team:Bielefeld-CeBiTec/siRNA">here.</a>
In our project we introduce RNA interference (RNAi) and transcription repression with small interfering (si)RNAs as an alternative to CRISPR/Cas. To use siRNA as silencing agents for the gene-of -interest we propose a two-step design process. At first potential siRNAs for prokaryotic organisms must be designed. In the second step the silencing effect of these siRNAs can be validated by our siRNA vector system Tace. To facilitate the initial siRNA design step, we developed a siRNA construction tool, which can find possible siRNAs for a given gene sequence and calculate their gene silencing probability. It consists of the three modules siRNAs for RNAi, siRNA, and check siRNA. Obtained siRNAs are perfectly compatible with our siRNA vector system. To the best of our knowledge, this is the first tool dedicated to predicting customized siRNA for the application in prokaryotes. This Python tool comes in two versions: a command line application and an easy-to-use graphical interface.
siRNAS short introduction
siRNAs are small single- or double-stranded RNAs with an average length of 21-25 nucleotides. They are non-coding RNAs which can bind a specific complementary coding mRNA and silence its function. During eukaryotic RNAi siRNAs are loaded to Argonaute proteins, which carry out the repression, either by blocking mRNA translation or by degrading the mRNA (Siomi and Siomi, 2009). More detailed information on both possible siRNAs mechanisms are found here.
Choosing appropriate design methods
In 2012 the iGEM team SYSU-Software integrated an siRNA cDNA designer as a small part in their project. siRNAs designed with this tool were applicable in eukaryotic organisms. They included two different design methods: Tom Tuschl’s method and Rational siRNA design.
Figure 1: Structure of an siRNA designed with Tom Tuschl's method. Both siRNA have a characteristic 'TT' overhang at the 3'-terminus.
Tom Tuschl’s method focuses mainly on the existence of 5’ and 3’ ‘TT’ overhangs (Figure X)(Elbashir et al., 2001). These are not compatible with overhangs and scaffold sequences necessary for the prokaryotic mechanisms. Therefore, we decided to use the Ui-Tei rules as an alternative design method (Naito and Ui-Tei, 2012). Furthermore, we adapted the Rational siRNA design since it was more suitable for our application (Reynolds et al., 2004). Both design rules apply only to the 19nt long target binding sequence.
Rational siRNA design
By a systematic analysis of 180 eukaryotic siRNAs Reynolds et al. identified eight criteria that are important for ther functionality (Reynolds et al., 2004). Each criterion gets a sciore that can be either positive or negative, corresponding to its effect on the siRNA. All siRNA candidates that have a score above six are potential high functional siRNAs.
Table 1:Rational siRNA design criteria with corresponding score
Rule
Score
30%-52% G/C content
+1
At least 3 'A/U' bases at positions 15-19
+1 (for each 'A/U' base)
Absence of internal repeats (\(T_m \lt 20\))
+1
An 'A' base at position 3
+1
An 'A' base at position 19
+1
An 'U' base at position 19
+1
A base other than 'G' or 'C' at 19
-1
A base other than 'G' at position 13
-1
The melting Temperature Tm is calculated as followed (Kibbe, 2007):
$$ T_m = 79.8 + (18.5 * log_{10}[Na^+]) + (58.4 * [\text{G/C content}]) \\+ (11.8 * [\text{G/C content}]^2) - \left(\frac{820}{\text{[G/C content]}}\right)$$
Ui-Tei rule
Ui-Tei et al analyzed 62 eukaryotic siRNAs and identified four design rules for effective siRNAs (Ui-Tei, 2004). Only siRNAs fulfilling all four criteria are considered functional siRNAs.
An ‘A’ or ‘T’ at position 19
A ‘G’ or ‘C’ at position 1
At least five ‘U’ or ‘A’ residues from positions 13 to 19
No ‘GC’ stretch more than 9nt long
Calculating silencing probability
Not only the sequences of possible effective siRNAs are to be determined and returned by the tool, but also the probability with which they are effective. This probability can be calculated with the help of Bayes’ theorem by calculating probabilities of dependent events. The following calculations and formular are based on Takasaki (2009).
The initial hypothesis is that the given siRNA effectively silences an mRNA. To perform the calculations a prior probability is necessary. The prior probability for effective gene silencing of mammalian genes can be obtained from former siRNA experiments and is approximately 0.1 (Takasaki, 2009). Since we have no data on prokaryotic siRNAs, we use the same prior probability for our prediction.
The gene silencing probability can be described as:
$$ P(eff|X) = \frac{P^{eff} P(X|eff)}{P(X)} \qquad (1)$$
\(P^{eff}\) is the prior probability 0.1 as mentioned above. The siRNA sequence is represented by \(X\), where \(X_1, X_2 ... X_n\) belong to the possible nucleotides adenine, guanine, cytosine and thymine. As \(P(X|eff)\) is the probability, that the given siRNA sequence will effectively silence if the nucleotides belong to the frequent nucleotides of common effective siRNAs, it is computed as the product of the probabilities that a particular nucleotide is located at a particular position of the siRNA:
$$ P(X|eff) = \prod_{i=1}^{19} q_{x_i^n}^{eff} \qquad (2)$$
\(q_{x_i^n}^{eff}\) indicates how likely the occurrence of base \(n\) is at position \(i\) based on known effective siRNAs. It can also be called frequency ratio of \(n\) at position \(i\). The last element \(P(X)\) of formula \((1)\) is the possibility that \(X\) will effectively silence the target sequence. It is the sum of the probability that \(X\) is effective if its nucleotides are found in effective siRNAs plus the probability that \(X\) is effective if its nucleotides are found in ineffective siRNAs. Both probabilities are weighted with the prior probabilities \(P^{eff}\) and \(P^{inf} = 1-P^{eff}\).
$$ P(X) = P^{eff} P(X|eff)+P^{inf} P(X|inf) \qquad (3)$$
\(P(X|inf)\) is calculated similar to \(P(X|eff)\) and is the probability that \(X\) will effectively silence if the nucleotides belong to the frequent nucleotides of common ineffective siRNAs.
$$ P(X|inf) = \prod_{i=1}^{19} q_{x_i^n}^{inf} \qquad (4)$$
In this case, \(q_{x_i^n}^{eff}\) indicates how likely the occurrence of base \(n\) is at position \(i\) based on known ineffective siRNAs.
With all defined formulas \((2)\),\((3)\) and \((4)\), formula \((1)\) can now be calculated as follows:
$$P(eff|X) = \frac{P^{eff} P(X|eff)}{P^{eff} P(X|eff)+P^{inf} P(X|inf)} \\\\= \frac{P^{eff} \prod_{i=1}^{19} q_{x_i^n}^{eff}}{P^{eff} \prod_{i=1}^{19} q_{x_i^n}^{eff}+P^{inf} \prod_{i=1}^{19} q_{x_i^n}^{inf}} $$
In order to actually calculate the silencing probability, only the frequency ratios \(q_{x_i^n}^{eff}\) and \(q_{x_i^n}^{inf}\) of the individual nucleotides at positions 1 to 19 are missing. These could be taken from the same publication from Takasaki as the calculations.
For the frequency ratios 833 effective and 847 ineffective siRNAs from previous publications were analyzed. For each nucleotide, the probability of occurrence was determined for each position of the siRNA. Different models were taken into account in the calculation. First of all, the occurrence of the different nucleotides at positions 1 to 19 can be considered independently. The probabilities for each position are then calculated independently. However, the occurrences of the nucleotides can also be considered dependently. This means the occurrence of a nucleotide depends on the nucleotide at the position before. For the calculation of dependent probabilities, the Simple Markow Model was used. It has been found that the resulting silence probability is most accurate when the frequency ratios of the effective siRNAs are calculated dependent and the frequency ratios of the ineffective siRNAs are calculated independent. All frequency ratios can be looked up here.
Together with the frequency ratios it is now possible to calculate the silencing probability for the 19 bp long binding site of siRNAs.
siRNA overhangs and scaffolds
In order to achieve effective gene silencing or knockdown, the 19 nt binding sequence must be supplemented with overhangs. There are different sequences that can be added to the binding sequence for different functionalities. Figure 2: Possible siRNA constructs for RNAi.
In Figure 2, the scheme of the siRNAs after cunstruction is shown. To trigger the mRNA degradation by the RNase E, the 5’-terminal triphosphate of the siRNA is converted to a monophosphate by the RNA pyrophosphohydrolase (RppH). For the siRNA to be recognized by the RppH, the 5’ end of the siRNA have to start with the nucleotides adenine and guanine. Furthermore, the nucleotides at position three and four are not allowed to match with the target mRNA(Foley et al., 2015). At the 3’ end of the siRNA the small MicC scaffold is added, which facilitates the hybridization of siRNA and target mRNA and protects the siRNA from degradation (Na et al., 2013).
Figure 3: Possible siRNA constructs for gene silencing.
Figure 3 shows the scheme of a siRNA that should only silence the mRNA target. To achieve a higher stability of the siRNA, the outer membrane protein (Omp) A scaffold is added at the 5’ end. In addition, the hybridization of the siRNA and the target mRNA should be facilitated by MicC again.
These overhang and scaffold sequences are also part of our vector system. If our vector system is selected when using our tool, the fitting overlaps to our vectors are added automatically. More theoretical information about the overhangs and scaffolds can be found here.
Check siRNA
Beside the construction of siRNAs, we also implemented a check siRNA functionality. For a given target sequence and a corresponding siRNA it is checked whether the siRNA might bind to its target and how well it fulfills the described criteria’s. Furthermore, its silence effectivity is calculated.
Command line application
The command line application can be obtained directly here or downloaded from our GitHub repository. For the execution of this command line application Python 2.7 needs to be installed.
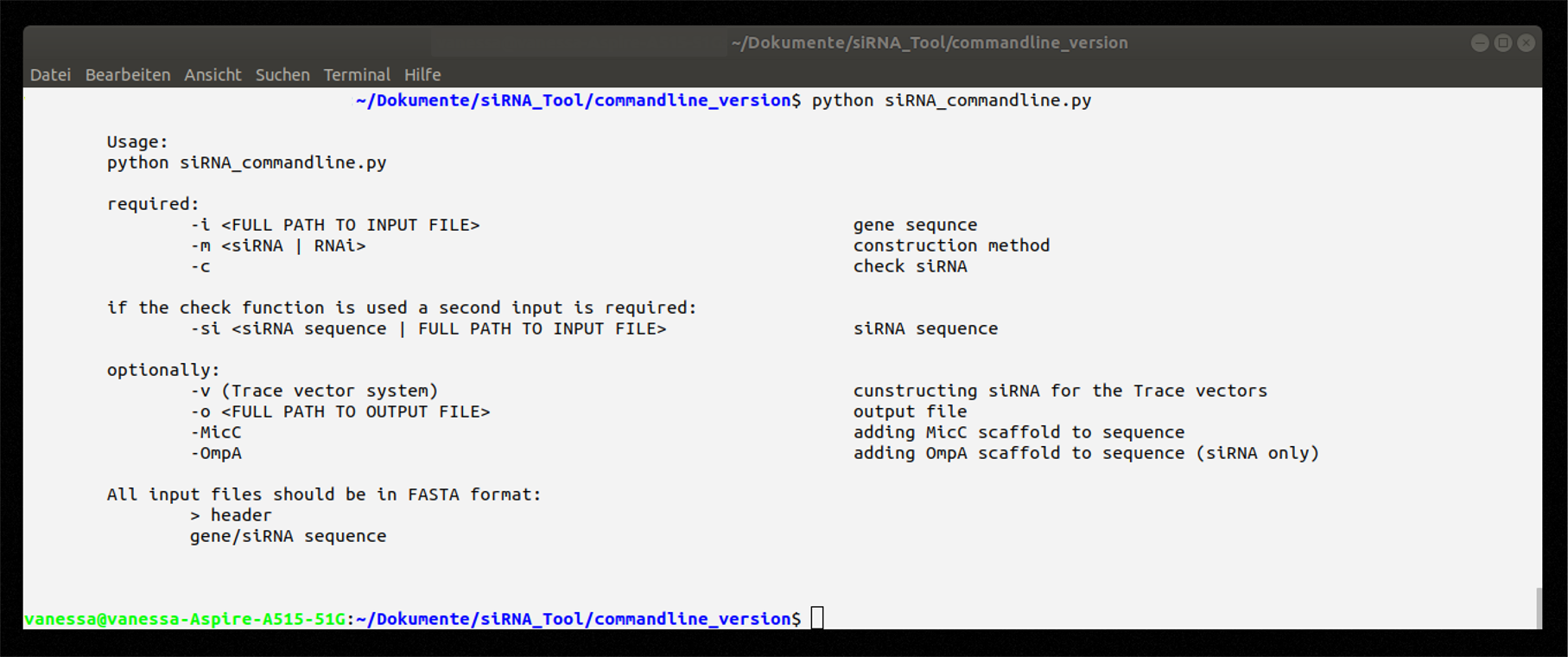
Figure 4: Instruction on how to use the command line application.
An overview of the necessary and optional arguments gives Figure 4. For more information a README is available in our repository. Figure X shows the execution of the tool with a GFP gene sequence.
All resulting siRNAs are saved in one FASTA file. This simplifies the integration into different workflows. For example, it is possible to test the siRNAs on off-target bindings site using Blast. An exemplary call of the application can be seen in Figure 5.
Figure 5: Exemplaric call of the command line application.
Graphical Interface usage
As the command line application, the graphical interface version can either be downloaded directly here, or via our GitHub repository.
In the graphical interface, the modules are divided into different tabs (Figure 6). The last tab contains usage and copyright information.
Figure 6: The different modules are diveded into different tabs.
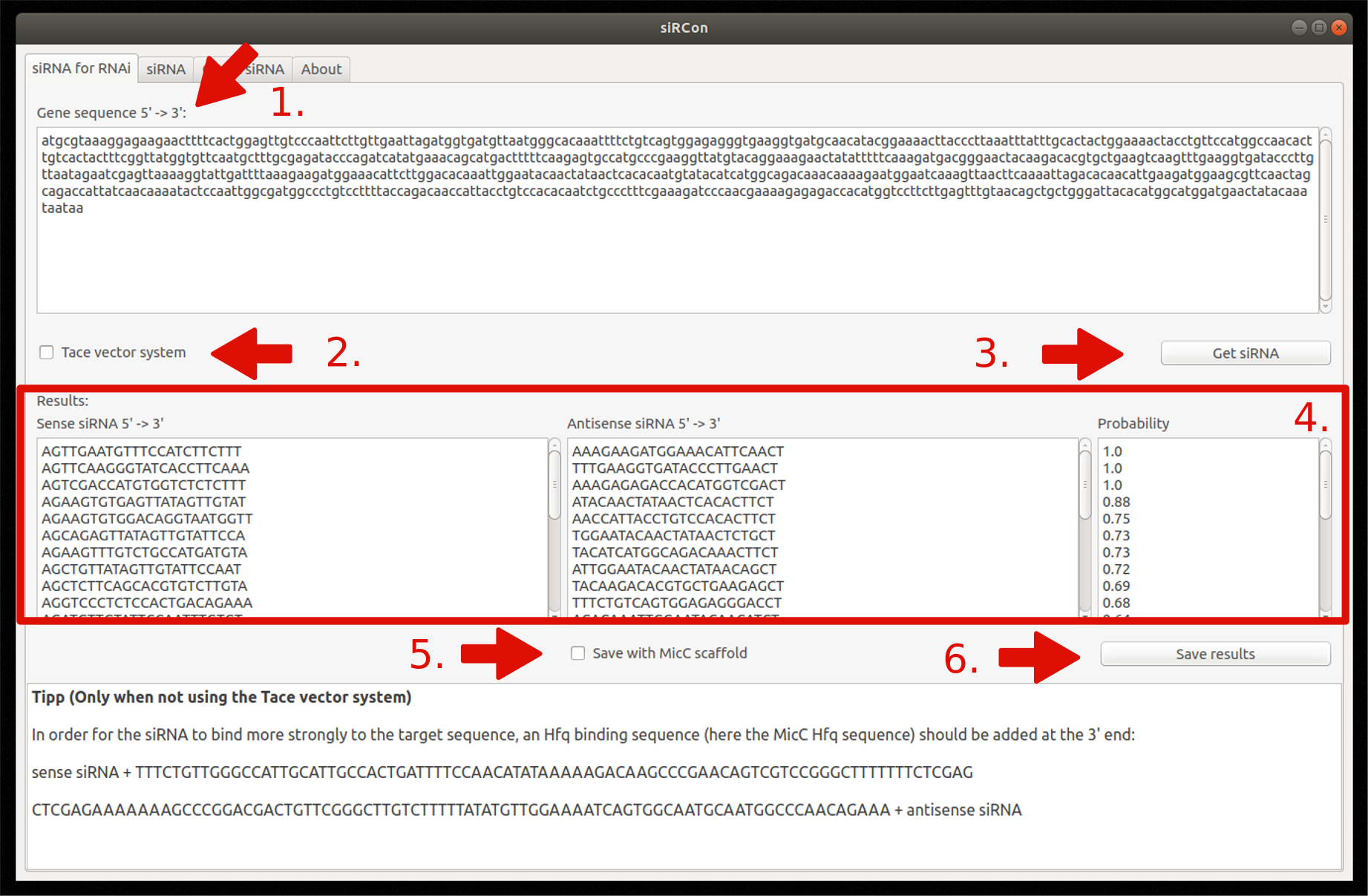
1. siRNA for RNAi
Insert gene sequence
Choose Tace vector system (optionally)
Constructions of siRNAs
View resulting siRNAs (sense and antisense sequence) and their corresponding probability
Decide if siRNAs should be saved with MicC scaffold (only if Tace is not used)
Save results as FASTA file
Figure 7: Overview and steps of the siRNA for RNAi module.
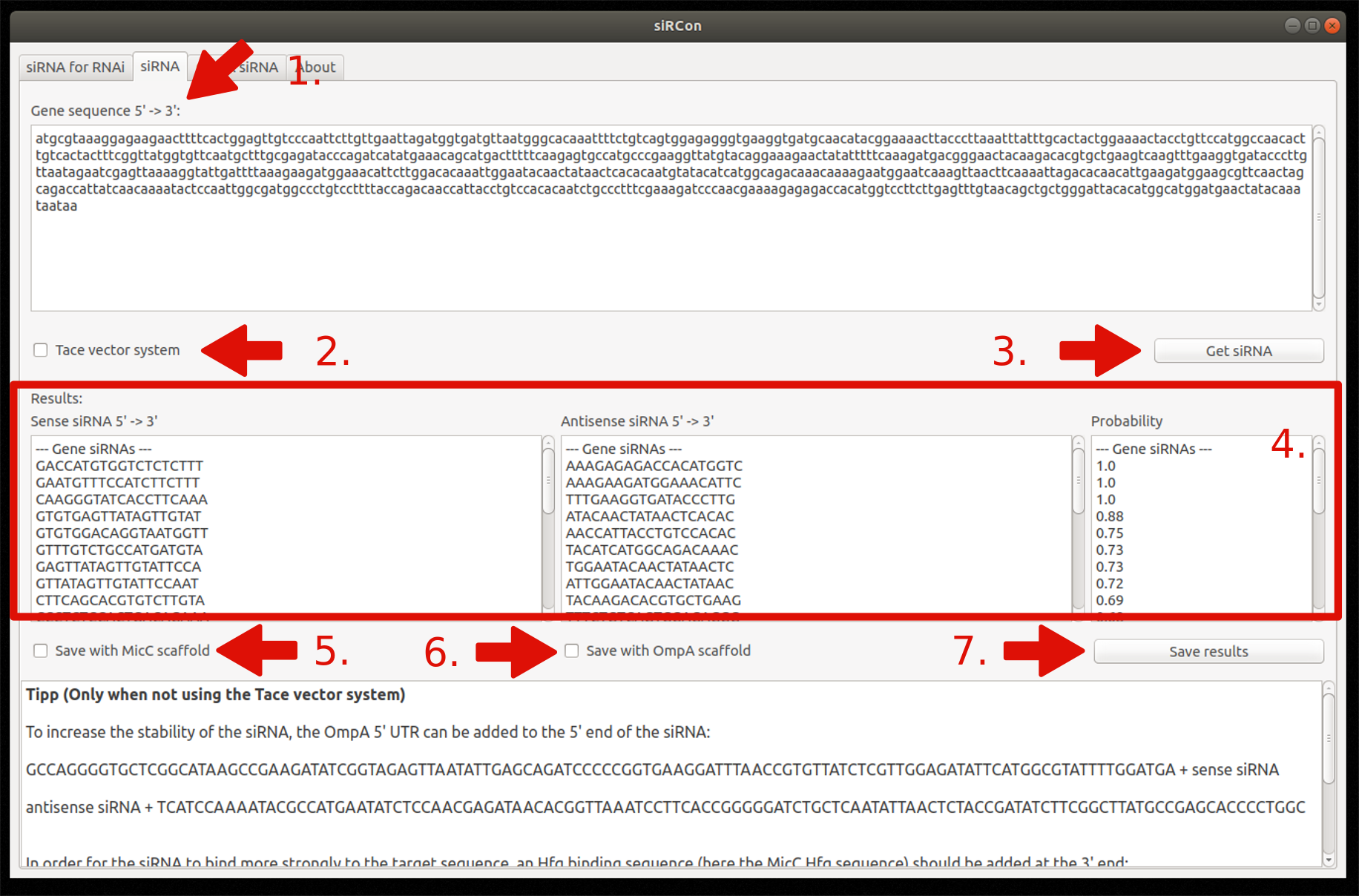
2. siRNA for silencing
Insert gene sequence
Choose Tace vector system (optionally)
Constructions of siRNAs
View resulting siRNAs (sense and antisense sequence) and their corresponding probability
Decide if siRNAs should be saved with MicC scaffold (only if Tace is not used)
Decide if siRNAs should be saved with OmpA scaffold (only if Tace is not used)
Save results as FASTA file
Figure 8: Overview and steps of the siRNA for silencing module.
3. Check siRNA
Insert gene sequence
Insert siRNA sequences
Choose method the siRNA was constructed for (siRNA for RNAi or siRNA for silencing)
Choose if siRNA was constructed for Tace (optionally)
Validation of entered siRNA for given target gene sequences
View results
Save results (optionally)
Figure 9: Overview and steps of the check siRNA module.
Outlook
To help future iGEM teams to control gene expression, we developed siRCon, a bioinformatic application for generation of high-fidelity siRNA sequences in prokaryotic organisms. We introduce this method as an alternative to CRISPR/Cas, since it is open source and free of charge.
In the future, further improvements and extensions of this applications are intended. On the one side, eukaryotic siRNAs will also be constructed. This is how we want to provide a universal tool for siRNAs. On the other side, we want to improve the already existing features, especially the check siRNA functionality.
Elbashir, S.M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498. Foley, P.L., Hsieh, P., Luciano, D.J., and Belasco, J.G. (2015). Specificity and evolutionary conservation of the Escherichia coli RNA pyrophosphohydrolase RppH. J. Biol. Chem. 290: 9478–9486. Kibbe, W.A. (2007). OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res 35: W43–W46. Na, D., Yoo, S.M., Chung, H., Park, H., Park, J.H., and Lee, S.Y. (2013). Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 31: 170–174. Naito, Y. and Ui-Tei, K. (2012). siRNA Design Software for a Target Gene-Specific RNA Interference. Front Genet 3. Reynolds, A., Leake, D., Boese, Q., Scaringe, S., Marshall, W.S., and Khvorova, A. (2004). Rational siRNA design for RNA interference. Nature Biotechnology 22: 326–330. Siomi, H. and Siomi, M.C. (2009). On the road to reading the RNA-interference code. Nature 457: 396–404. Takasaki, S. (2009). Selecting effective siRNA target sequences by using Bayes’ theorem. Computational Biology and Chemistry 33: 368–372.