<b>Figure 7: 200kV TEM image with scale bars of a close up measurement of NP formed inside a href="http://parts.igem.org/Part:BBa_K2638612">mutant ferritin</a>.
+
<b>Figure 7: 200kV TEM image with scale bars of a close up measurement of NP formed inside a< href="http://parts.igem.org/Part:BBa_K2638612">mutant ferritin</a>.
For our project we used two different types of ferritin, the bacterioferritin BfBr and the human ferritin. For our purposes it was enough to only use the human ferritin heavy chain subunit which when assembled forms a ferritin cage named HuHF. We showed increased iron nanoparticle formation through BfBr overexpression and engineered HuHF to investigate stability changes in the presence of copper ions. Furthermore, we constructed HuHF mutants with the ability to form gold and silver nanoparticle.
Enhanced Stability and Reassembly of Mutant Ferritin
We chose E. coli as the host organism for producing of a stability enhanced mutant of HuHF heavy chain. Wild type ferritin consists of two different subunits, the heavy and the light chain but for simplicity reasons we only use the heavy chain which can form the ferritin cage on its own without the light chain being required.
We used DNA synthesis from IDT based on the part BBa_K1189019 which was introduced by Calgary 2013 but codon optimized the sequence for expression in E. coli resulting in the following part.
Using the synthesized sequence of the human ferritin as a template we introduced the desired mutations via primers.
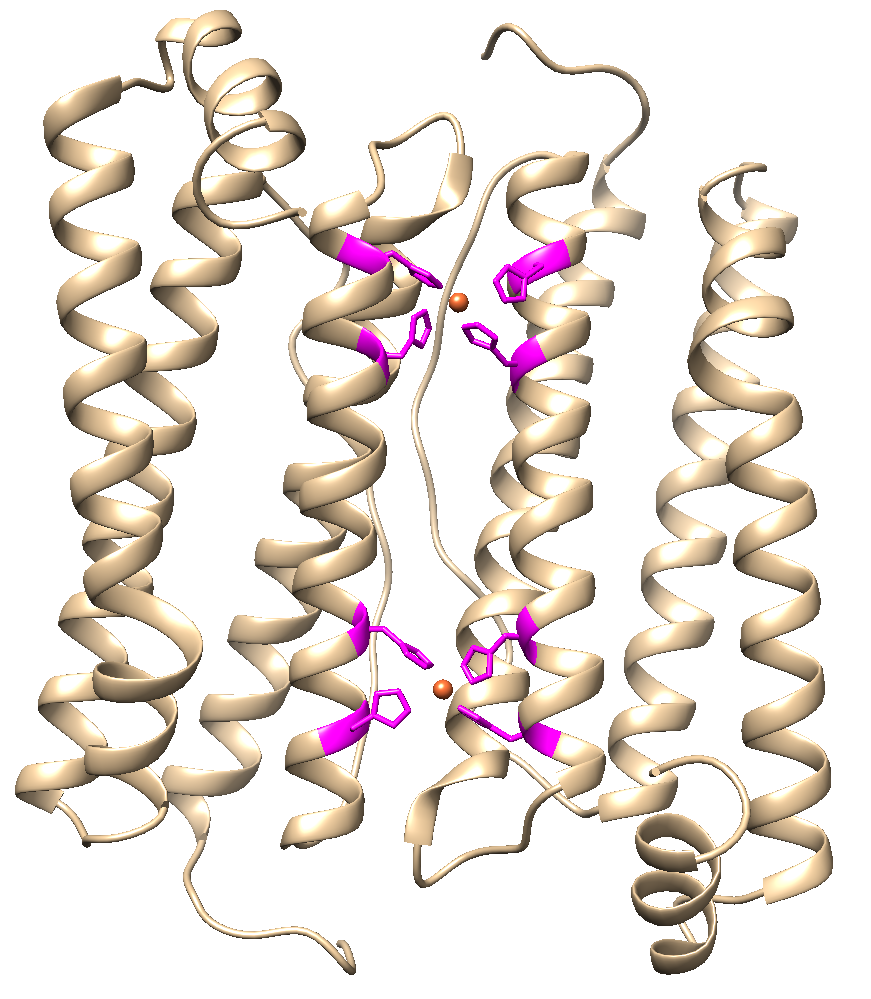
We changed 4 amino acid to histidine which resulted in two new binding regions for copper in the surface area between two adjacent subunits (Figure 1) which supposedly (Wang Z. 2018) enhances binding strength between the subunits and resulted in the following part BBa_K2638601.
Figure 1: Four amino acid changes should lead to enhanced stability after copper ions are added to the synthesized HuHF.
The second mutation we introduced was meant to weaken the ferritin cage so it can be disassembled more easily. By shortening the C-terminus of the ferritin subunit removing two structural parts the E-helix and the DE turn (see Figure 2) the resulting part BBa_K2638602 should disassemble at a less destructive pH of below 4 instead of being below 2 for wild type human ferritin (Chen H. 2016).
Figure 2: Wild type ferritin with the E-helix and DE turn colored in green.
We then combined both mutations into one part BBa_K2638603. We had the hope to end up with a ferritin version that combines booth strengths being easily assembled and filled with a payload of interest and made stable again by adding copper ions afterwards.
To test if the different mutant ferritin variants show the desired properties we planned to disassemble the different ferritin mutants into single subunits first by changing the pH as described in the literature (Chen H. 2016). After the disassembly step we would mix two different ferritin subunits for example one with a mTagBFP tag BBa_K2638610 and one with a His Tag.
The His Tag carring ferritin BBa_K2638614 could bind to the his purification column while the mTagBFP tagged subunit could not bind to the column on its own. Only if the his and mTagBFP tagged ferritin subunits successfully disassembled and after mixing them and bringing the pH back to normal reassembled, would we expect to see his purification columns that fluorescent.
All the mutations to the ferritin were introduced by primers binding to the synthetized HuHF sequence and the resulting PCR products were combined via Gippson Assembly.
We used primers either binding to the synthesized HuFH, the mTagBfp or the ends of the vector to introduce overhangs for adding the linker, the his tag and the mTagBFP to the ferritin heavy chain. The linker was introduced to keep the mTagBFP from being too close to the ferritin subunit possibly resulting in steric hindrance. Steric hindrance could influence the assembly process of our mutant ferritin. mTagBFP and linker where attached to the N-terminus of the ferritin since the N-terminus is positioned on the outside of the ferritin cage.
For protein purification we choose a commercial intein tag which cleaves itself of after protein purification was performed only leaving back the desired protein (see Intein IMPACT protocol).
The resulting PCR product was inserted via Gipson Assembly into pTYB11. pTYB11 is an commercially available protein purification vector which contains the intein tag which cleaves itself of during the protein purification process. Using the intein purification tag has the advantage over his tags that during purification the tag detaches itself and stays behind bond to the column while the protein of interest gets released. This way we could produce ferritin mutants with different functional proteins on the outside and don't had to worry that the tag influences the function of our mTagBFP for example.
pTYB11 was than transformed into E. coli Dh5α for high copy plasmid production. Plasmid iso was performed and the plasmids were retransformed into the protein expression strain E. coli ER2566.
Correct Folding And Assembly Of Mutant Ferritin
Using the impact protein purification protocol, we were able to purify our blue fluorescent ferritin shown in figure 3 the top right of the picture.
Figure 3: Top right ferritin with fused mTagBFP glowing blue under UV light indicating successful expression of fusion protein. Just behind it glowing red the mRFP tagged ferritin mutant containing 4 copper changes and also missing the E-helix and DE turn.
We also performed a fluorescence scan using the Tecan reader to validate what could be seen under UV light with the naked eye. Excitation and emission values specified for mTagBFP where in agreement with our measurements.
SDS page with the purified mTagBFP tagged ferritin was performed to validate the promising results we achieved with the fluorescence scan using the Tecan reader. The gel shows a band at around 45 kDa which agrees with the size prediction of the modified ferritin subunit (1236 bp ; 45,7 kDa).
Figure 4: SDS gel shows the right bands indicating successful expression of our fusion ferritin.
Interestingly the SDS gel might also be used as an indicator for successful mutant ferritin formation.
Because the ferritin is a large round cage only a subset of subunits which have an intein tag attached to them should interact with the column allowing the rest of the intein tags to stay unbound and be released after cleavage.
The intein purification tag has a size of 55 kDa and should therefore be present as a band above the fusion protein when the column is fully loaded giving us a direct indicator for successful ferritin formation.
Since such an intein band was not present we concluded that the chitin column was not overloaded and had enough free space for the released intein tags to bind to.
To make sure that the mutant ferritin-mTagBFP BBa_K2638612 assembled correctly protein expression and purification was repeated in media enriched with 100 mM of Iron(III) citrate (Iron(III) citrate was boiled in LB to dissolve crystals). The E. coli strain ER2566 was used for intein tagged protein expression. Using iron enriched medium the formation of detectable iron nanoparticles was expected only if the mutant ferritin has successfully assembled inside the cells and kept its functionality in regards to forming correctly and binding as well as oxidising iron ions. Despite the introduction of the mutations (removing the E-helix and DE turn) correct assembly could be proofed this way.
The iron loaded ferritin was again purified following the intein purification protocol and the presence of the mutant ferritin was confirmed by fluorescence measurements as described above. Afterwards we examined the mTagBFP tagged NP using a small 80kV desktop transmission electron microscope (TEM).
Figure 5: Size of our ferritin modeled using Chimera.
The TEM images seemed to confirm the formation of iron nanoparticles suggesting that the ferritin cage had formed correctly inside the cell and also keeped its function to take up iron.
It should be noted however that the resolution of the 80kV TEM used for making these images is not ideal and we cannot say with full certainty that the structures with a size of ~29 nm visible in the TEM images are the mutant ferritin. To the best of our knowledge nothing else could be responsible for creating such uniform structures visible under the TEM.
As a side note we should point out that proteins are in general not visible well in the electron microscope but we noted a clearly visible white circle around a darker spot which might be caused by the protein shell around the iron core.
The iron core however should be clearly visible as a dark spot in the TEM images. If the resolution of the TEM is good enough it is even possible to see the uniform metal structure of the iron core like we showed in a previous experiment.
Figure 6: 80kV TEM image with scale bars measuring ferritin nanoparticles.
To make a definitive conclusion if an iron NP core can form in our mutant ferritin BBa_K2638612 we went to a higher resolution TEM performing close up images of the iron core. The iron core resolution is enhanced but contrast is weaker than the images acquired by the 80 kV TEM due to the 200kV TEM used for making those images having a higher energy beam. A second factor which influences the visibility in the TEM images is the composition of the metal core. Fe(III) oxide shown under the TEM is less visible compared to elemental metal. How elemental metal looks under the TEM can be seen on our page on gold and silver forming ferritin.
To confirm that the particles consist of iron we performed an EDX measurement using the TEM. EDX measurements where however not precise enough to give a conclusive answer about the composition of the metal core. The electron beam is bigger than the size of the 8 nm metal core therefore it is not possible to measure a single NP directly but only large clusters of NP. Measuring large clusters has the disadvantage of not being able to see the exact shape and size of the NP which could lead to wrong conclusions when other particles than the one formed inside the ferritin are being measured. The precise size of 8nm or less and the round shape are the best indicators for NP formed by ferritin.
Figure 7: 200kV TEM image with scale bars of a close up measurement of NP formed inside a< href="http://parts.igem.org/Part:BBa_K2638612">mutant ferritin.
Analysis of Enclosed Fluorophores by Fluorescence Correlation Spectroscopy (FCS) using Zeiss LSM780
To test if our modified ferritin can be used as a cage to enclose various proteins and molecules of interest we attempted to enclose mRFP (27 kDa) and the much smaller fluorescein (0.33 kDa) respectively.
mRFP was overexpressed in psB3t5, the cells where ribolysed and purified using a 0.5mL protein filter (Amicon Ultra). Synthetized fluorescein was used which our team member Jakob Zubek also synthetized himself in one of his chemistry practical courses.
To compare the enclosure capability we used the wild type HuHF and compared it to our modified ferritin missing the e-helix and DE turn (BBa_K2638612).
As the objective of this experiment was to look for differences in the pH values necessary for disassembly of the unmodified ferritin and our modified ferritin we exposed the ferritins to different pH conditions. In the literature the pH threshold for disassembly is 2.10 for the ferritin wild type and 4 for our ferritin variant (Chen H. 2016). Reassembly should occur at a pH of around 7.5.
We therefore exposed the wild type and the mutant ferritin to a pH of below 1 and to roughly 3 using HCL. Afterwards we added purified mRFP or fluorescein in the highest possible concentration so that passive enclosure could take place. After returning the pH back to 8 we characterized the probe via FCS.
Initially we wanted to look directly for overlapping fluorescence signals of the BFP tagged ferritin (BBa_K592100) and the mRFP enclosed inside. But the effects of brownian motion and the small size (~29 nm) of our fusion ferritin makes this kind of visualization impractical, which is why we turned to fluorescence correlation spectroscopy (FCS) as an alternative.
FCS uses a laser to excite single fluorescent molecules passing in front of it through a very small volume. A detector then measures the intensity and time of the fluorescent signal which can be used to estimate the mass and number of molecules in front of the laser.
Since free fluorophores are much smaller than ferritin encapsulated fluorophores they move through the small detection volume at a much higher speed. Thus the two populations of small free fluorophores and larger slower ferritin-associated fluorophores should be discernable by FCS.
By measuring the individual times of the passing particles and by taking the average we received a table of times which describe how long the fluorophores spend in front of the laser.
Unfortunately, the measured times did not confirm our assumptions. We attribute this to the contamination with other large proteins who might also interact with our fluorophores changing their average speed despite not being enclosed.
We expected to see very short average times for the fluorescein on its own which could be confirmed by measuring an average time of 0,005 µs.
Measuring the speed of fluorescein after opening and closing the ferritin cage via lowering and raising the pH we saw a significant slow down but we could not definitively say that our engineered ferritin is responsible or if the fluorescein interacts with other impurities in the solution. Since our negative control containing a protein mix which should not enclose any fluorescein we decided to not use the data for the characterisation of our mutant ferritin.
Our TEM images (Figure 7) showed that many impurities are mixed into our eluate and we therefore hypothesize that those impurities influence the speed of our fluorophores significantly resulting in the highly different speeds observed by our measurements.
Human Ferritin engineering
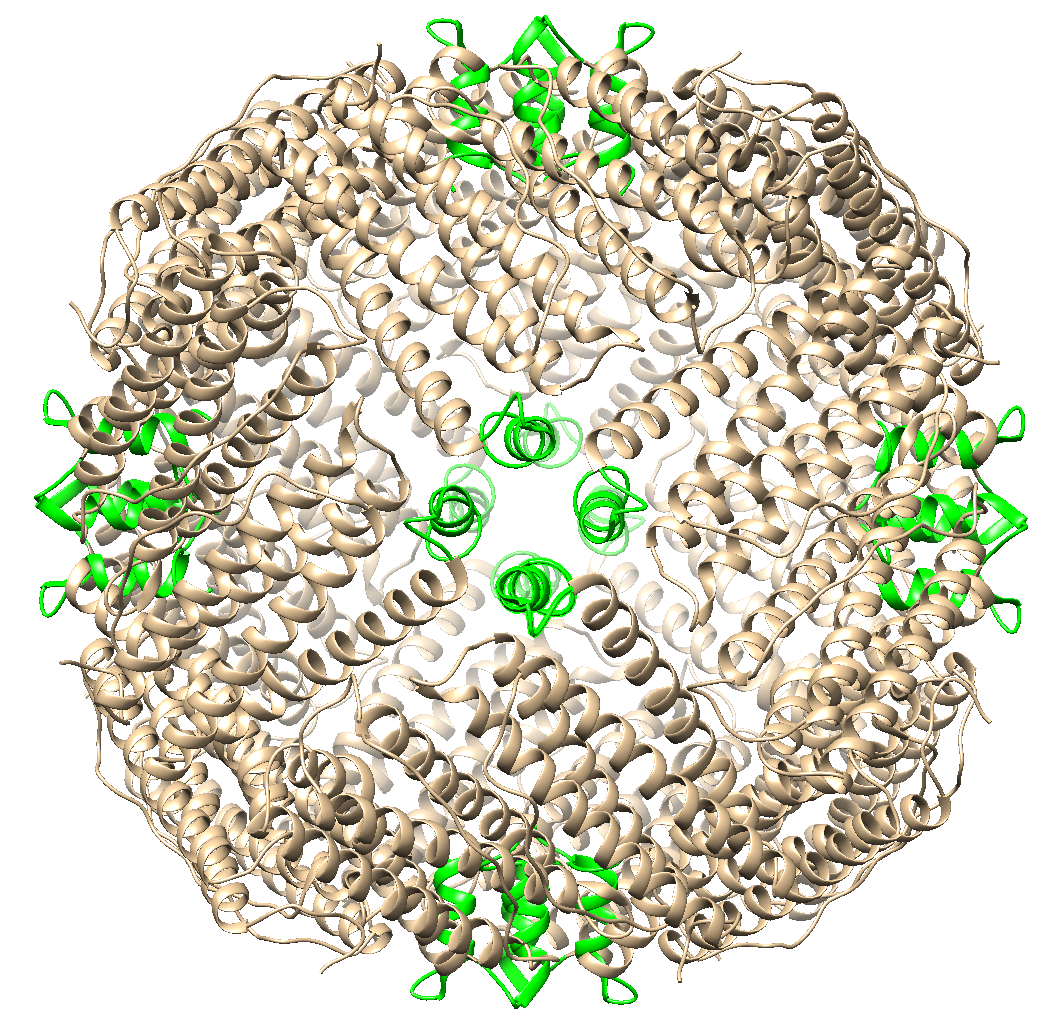
To engineer ferritin to build silver and gold nanoparticles, we changed certain amino acids residues in the human ferritin. The results can be found on our Part Improvement page.
Figure 8: Multiple human ferritin subunits with the introduced amino acid changes marked in red. The amino acid modifications change the outer and inner surface of the ferritin cage allowing for the storage of gold and silver ions primarily on the inside.
Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. Butts, C.A., Swift, J., Kang, S., Di Costanzo, L., Christianson, D.W., Saven, J.G., and Dmochowski, I.J. (2008).. Directing Noble Metal Ion Chemistry within a Designed Ferritin Protein † , ‡. Biochemistry 47: 12729–12739.
Castro, L., Blázquez, M.L., Muñoz, J., González, F., and Ballester, A. (2014).. Mechanism and Applications of Metal Nanoparticles Prepared by Bio-Mediated Process. Rev. Adv. Sci. Eng. 3.
Ensign, D., Young, M., and Douglas, T. (2004).. Photocatalytic synthesis of copper colloids from CuII by the ferrihydrite core of ferritin. Inorg. Chem. 43: 3441–3446.
Goujon, M., McWilliam, H., Li, W., Valentin, F., Squizzato, S., Paern, J., and Lopez, R. (2010).. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38: W695-699.
Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C., and Ferrin, T.E. (2004).UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612.
Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J., Thompson, J.D., and Higgins, D.G. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7: 539.
Ummartyotin, S., Bunnak, N., Juntaro, J., Sain, M., and Manuspiya, H. (2012). . DSynthesis of colloidal silver nanoparticles for printed electronics. /data/revues/16310748/v15i6/S1631074812000549/.
Wang, L., Hu, C., and Shao, L. (2017a).. The antimicrobial activity of nanoparticles: present situation and prospects for the future. Int. J. Nanomedicine 12: 1227–1249.
Wang, Z., Gao, H., Zhang, Y., Liu, G., Niu, G., and Chen, X. (2017b).. Functional ferritin nanoparticles for biomedical applications. Front. Chem. Sci. Eng. 11: 633–646.
Wang, Z., Dai, Y., Wang, Z., Jacobson, O., Zhang, F., Yung, B. C., ... & Chen, X. (2018).. Metal ion assisted interface re-engineering of a ferritin nanocage for enhanced biofunctions and cancer therapy. Nanoscale, 10(3), 1135-1144.
Chen, H., S. Zhang, C. Xu, and G. Zhao. (2016). "Engineering protein interfaces yields ferritin disassembly and reassembly under benign experimental conditions." Chemical Communications 52, no. 46 : 7402-7405.