However beautiful the strategy, you should occasionally look at the results
-- Winston Churchill
Initially, we tried to reproduce the experiment carried out by Beyer et al. (2015), to turn a sfGFP into a blue fluorescent protein by changing a single amino acid (Y66H). We could observe blue colonies and sequenced the plasmids to confirm the desired mutation. An alignment of the chromatogram is shown in figure 1.

The site directed mutagenesis performed with Aquaclonging (Y66H) was confirmed by sequencign for both tested clones. A tyrosin codon was changed to a histidine codon.
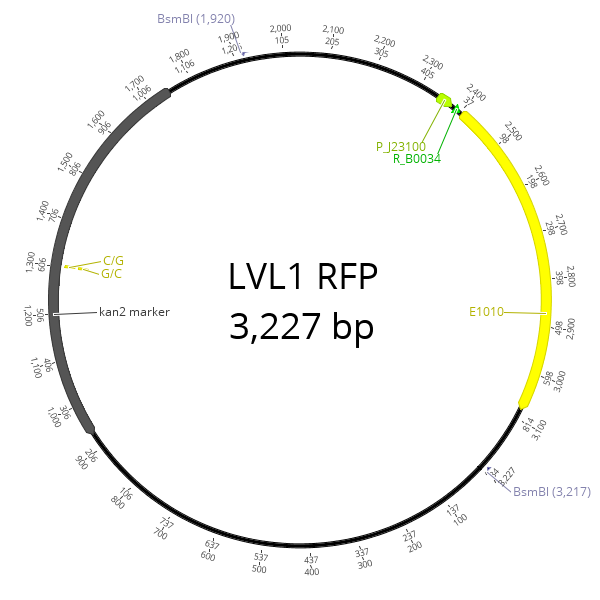
The experiment was designed such that correctly assembled plasmids result in RFP expressing V. natriegens colonies. The sfGFP CDS of pYTK, the plasmid used in the previous experiment was replaced by a RFP CDS. The resulting agar plate is shown in figure 2.

A plate full of colonies with a correctly assembed plasmid
Due to the limited amount of time, we were not able to repeat the experiments with four and five fragments, but we are certain that optimizing transformation protocols, the amount and ratio of DNA fragments or the length of homologous flanks will result in successfully assembled plasmids even for this very challenging application.
Growth experiments

After reading papers about V. natriegens deciding to order the strain ATCC 14048, we were eager to make our first growth curves.
Because V. natriegens is a marine bacterium organism and from literature (et al), we knew that we had to supplement our standard LB media with an additional 1.5% NaCl.
Being inexperienced with the new organism, we only managed to get a doubling time slightly better than for E. coli. Not disheartened by this, we did some more research and found several points we wanted to try.We tested the following media, LB 2.5 NaCl, LB v2 salts and Brainheart infusion v2 salts.

v2 salts are: 204 mM NaCl, 4.2 mM KCl, 23.14mM MgCl2 Brainheart infusion can not be autoclaved together with the v2 salts. From this we learned that growth was most vigorous in BHIv2, closely followed by LBv2. So we decided to make LBv2 our standard media going forward. It is cheaper as well as just being standard LB media, supplemented after autoclaving with the three common salts NaCl, KCl and MgCl2.
Additionally, some aspects of handling could be improved. For instance reducing the time outside of the incubator for measuring, thereby avoiding cooling of the media, and using baffled flasks to improve aeration. Applying these, we managed to get much better results, even beating the reported doubling time of 9.8 min
(Eagon et al. 1962). The graph shows a growth curve of V. natriegens in BHIv2; V.natriegens in LBv2 and the fastest E.coli stain (NEB Turbo) in LB media.We achieved an incredible growth rate of 7 min. Also, using baffled flasks, our overnight cultures routinely reached OD's from 14 to over 16. By using enpresso capsules, which slowly release glucose into the media, we could even best that and reach staggering OD's of around 26.

Salt experiments
To find the perfect salt concentration for V. natriegens, we conducted new experiments using different salt concentrations ranging from standard LB media with no additional salt and then added NaCl in 1% steps, up to a concentration of 12 % in total.


Afterwards we incubated the cultures that showed no growth on media with the ideal concentration of 2.5% and let them recover to check if they were hibernating, but still alive after being exposed to the high salt concentration. Until 10% of total salt in the media, V. natriegens did not recover from growing in higher salt concentrations.
Antibiotic tolerance of Vibrio natriegens

The required concentrations for selection in V. natriegens is still unknown for most standard antibiotics. Since these were a prerequisite for our project, we strove to find them early on.
An easy method to get a good idea of the effective concentrations is to make agar plates with a gradient , then set spots of your culture along that gradient. (Figure 1)
We found that the penicillins ampicillin and penicillin, the arguably most used antibiotics, seemed to be completely ineffective at inhibiting V. natriegens . Fortunately, V. natriegens susceptible for the penicillin derivative carbenicillin. Since the carbenicillin resistance is mediated through the same enzyme as ampicillin resistance, the ß-lactamase, all constructs carrying this resistance can still be selected for.
The concentrations in the following table are working fine in our lab for selecting transformants with the resistance cassettes from our Toolbox.
|
WT ingibition [µg/mL] |
WT counter-selection [µg/mL] |
inhibition with resistance [µg/mL] |
|
|
Chloramphenicol |
2 |
2 |
<12 |
|
Carbenicillin |
50 |
200 |
600 |
|
Kanamycin |
65 |
200 |
400 |
|
Tetracycline* |
7 |
11 |
<12 |
|
Streptomycin |
80 |
/ |
/ |
|
Gentamycin |
30 |
/ |
/ |
|
Glycloserine |
<200 |
/ |
/ |
|
Penicillin |
<100 |
/ |
/ |
(Figure 2 Scheme of the most common antibiotics in synthetic biology and their relevant concentration in correlation with selection V. natriegens with and without a resistance cassette )
The first four antibiotics, Chloramphenicol, Carbenicillin, Kanamycin and Tetracycline are the antibiotics we commonly used in our project. For Golden Gate cloning we routinely switch between Chloramphenicol and Kanamycin. Because, it is highly sensitive to light, and we are using inducible promoters that are activated by a derivative of tetracycline and also reacting to the antibiotic, tetracycline only is used as our emergency antibiotic.

We used the pYTK plasmid from the Dueber Toolbox pYTK which has a GFP dropout for visual differentiation, as well as a Chloramphenicol resistance.
{kind=link}
We think the issue is that during the making and freezing of the competent cells, a lot of cells die, which releases the DNase from the wild type into the media. The DNases digest the DNA before it can get into the cells.
The reason why the transformation works without the knockout is that the DNases can't digest all 550 ng DNA and some is left over and can be taken up. You only get single colonies and this efficiency is too bad for cloning. The amount of DNA needed is too great for realistic applications and you would still need E. Coli for re-transformation. Since we think the DNase is the root of our problem, after we knock it out we want to give chemically competent cells and heat shock transformation another try. Next we tested the electroporation protocol, because it is reported to have a much higher efficiency. Akira Taket 1988 We made some minor adjustments to the protocol because we did not have the same equipment that was used in the paper and we wanted to use 3000xg instead of the rpm value for the centrifuge to make the protocol applicable to other labs.
The picture shows four plates of Vibrio natriegens , transformed with different concentrations of pYTK. Top left: 50ng; top right: 5ng; down below left: 0.5ng; down below right: 0.1ng. For better visualising the GFP the plates are lightened by UV light.
![]()
We used an electroporator from Eppendorf whose only variable parameter was the voltage. This protocol worked fine with 100 ng of DNA. But the efficiency still left something to be desired. Especially when we think about cloning, so we optimized the protocol and our handling again. We made sure that all buffers and centrifugation tubes were cooled to 4 °C when making competent cells before they reached an OD600nm of around 0.5 and that the samples were kept on ice to inhibit further growth.
We increased the centrifugation strength because after diluting the cells to a fix OD of 16, we produced 30 aliquots which isn't enough when you do triplicates. We also froze the cells in liquid nitrogen instead of in dry ice. For the electroporation we tested with different gap sizes, ranging from 1 to 2 mm. 1mm proved to work better for Vibrio natriegens. We preheat the recovery media to 50 °C because mixing the 4 °C cells in the chilled cuvette lowers the temperature of the media. Preheating the media to 37 °C would result in a temperature approximating 30 °C after pipetting and mixing with the cold cells. Afterwards we put the electroporated aliquots in a 37 °C water bath to help them recover while we are treating the other samples. More cells survive, therefore the efficiency increases.
In the paper of Weinstock et al. 2016, a voltage between 700 and 900V is recommended. One of our advisers said that a higher voltage increases efficiency so we tested voltages of 700V up to 1600V in steps of 100V.
The difference between 1600V and 900V was not noteworthy. It might be that with an increasing voltage (efficiency)also more cells die during the procedure, resulting in a consistent final efficiency. Electroporating 24 samples takes time and during the first and the last electroporation the first cells have about 15 min more time to recover, directly i a water bath. After applying these new changes the efficiency increased we are now able to successfully transform with concentrations as low as DNA of 0.1ng. Which is nearly what is achieved when working with E. Coli with heat shock. E. Coli still performs better in this, but we are working with the Vibrio natriegens wild type and it is already good enough for cloning.
Gibson cloning in Vibrio natriegens
The next step in our project is to test different cloning methods. A new and fast chassis needs a fast and clever cloning strategy. We wanted to show that the most commonly used cloning methods, Golden Gate cloning (Carola Engler et al. 2009)
and Gibson assembly
(Daniel G Gibson et al.
2009),

Consequently, we send sequenced samples of colonies showing red fluorescence and found all of them to harbor the correctly assembled plasmid without mutations. Cloning efficiency for Gibson assembly is usually higher than 90%, where very few cells still showed green fluorescence. ( Picture 4)

With this, we also prove that the efficiency of electroporation is high enough for Gibson cloning in V. natriegens. We found no significant difference when taking the plasmids from an E. coli preparation. After showing Gibson cloning with two fragments we wanted to check if we could do this method with up to five fragments. To test this we amplified pieces of the pYTK and RFP plasmids according to figure X and Y.
Using the same protocol as mentioned before, and also successfully demonstrated correct assembly, which we affirmed by sequencing.
While we had the Echo in our Lab, a machine for “pipetting” very low volumes, down to 2.5 nL, together we also tried doing Gibson assembly with it. The total volume of the mix was exactly one µL.
After assembling the samples in the 96 well plate and after transformation we got colonies for every cloning.
Again, we counted the cells to calculate the efficiency and again reached over 90% of correct transformants.



Golden Gate cloning and cloning in one day
Our Toolbox, the Marburg Collection is build for Golden Gate cloning and intended to be used in V. natriegens. Implementing this method, we want to enable combinatorial building of plasmids in which you can decide on the position for every part. To demonstrate that this method works, we devised an experiment, of building a Lvl2 strep tag.{kind=link}


{kind=link}
{kind=link}
{kind=link}
Cloning in Vibrio natriegens on the other hand is still much faster than cloning in E. coli, where you also have to struggle with the same problems. Weather you have a normal work day of eight hours or if you work like an iGEMer, you can save in each case a whole day by using Vibrio natriegens. That is immense. You can save third of the time by not waiting that long for cultures to grow. Additionally, there is still a lot of potential to build faster Vibrio natriegens strains. This could revolutionize cloning. All this combined with lab automatisation, clever machines doing the cloning steps, as we did with with the echo or the opentron ,performing heat shock transformations in 96 well plates and pick automatically after automatically plating the colonies. You could start your cloning on Friday, and as we are used to say in Marburg: “ Monday its finished”. This could be what accelerating synbio could look like.
Working with V. natriegens all the time, we also wanted to see it with our own eyes. To do that, we used several methods of transmission electron microscopy ( TEM ) and scanning electron microscopy (SEM).Our microtomes we made in two ways, by fixation in epoxy resin and in freezing to a grid. Thereby we can show you cuts through the cell (Picture 1-4 ) The method we also showed was platinum coal steaming the cells (Picture 5) and negative staining for visualising attachments like flagella.( Picture 6 and 7 )
On one picture we also have a three dimensional impression of the cells from scanning electron microscopy. (Picture 8 and 9 ) From the size of the pixels and the magnification you can calculate a scale bar, or directly measure the cells. What we can conclude from this is, Vibrio natriegens is about one to two µm long and has a diameter of around 1µm. It has a single polar flagellum and it looks like it is dividing along its equatorial axis. We could also make out something in the cell what could be storage compartments. ( Picture 10 )








At many different occasions, at meetups or conferences, we showed the growth curve of V. natriegens compared to E. coli. Other scientists were impressed by the extremely fast growth rate but even more by the high OD600 that we could show. We were asked many times if the high OD600 is really due to a high cell density or if it is rather caused by other components like cell debris or substances secreted to the medium which contribute to the measured absorbance.
To find out more about the growth dynamics, we decided to acquire a growth curve of V. natriegens in the most direct way, by counting cells in a flow cytometer. We inoculated three baffled flasks from stationary cultures and took samples in 15 minute intervals while the bacteria were incubated at 37 °C with shaking at 220 rpm. The OD600 of these samples was measured in a normal photometer and the cultures were then immediately analyzed by flow cytometry. The flow cytometer directs the samples through a thin capillary so that the cells pass a laser bean one by one and thus, can be counted and analyzed independently. A constant flow rate and time for data acquisition was set, which results in measuring a defined sample volume. Together with the counted events, the cells per volume can be calculated.

OD600 is shown in red and events/µL measured with flow cytometry is shown in blue
When we planned this experiment, we were most curious about the composition of the culture in the stationary phase to answer the question if the high OD that V. natriegens can reach is the result of a high cell density or if it can be traced back to other substances. Interestingly, both values, OD600 and events/µL start to stagnate at a similar time point (165 min). We interpret this result as a confirmation that the high OD is indeed caused by bacterial cells.
By carefully comparing the shape of both growth curves, we realized that, in fact, the most striking data in this plot can be found at the beginning of the experiment. While exponentially increasing values can be seen right from the start for the curve created from the OD600 data, a short lag phase is apparent when events/µL are plotted (figure 1). We tried to find an explanation for this observation and realized that the absorbance of a culture does not necessarily correlate with the concentration of cells but rather with the biomass inside the flask.

A single sample after 45 minutes is shown as an example. Each dot represents one event

The measured events/µL are shown in blue and on the left Y-axis. The forward scatter is displayed in green and on the right Y-axis
Taking all three datasets into account, we suggest that the cells start to grow in size upon provision of fresh medium but initially without undergoing cell division. This results in an increase volume of individual cells and thus, an increase of the measured OD600 but without significant changes to the cell concentration in the culture. After 45 minutes, when the forward scatter peaks, we assume that a majority of cells reach maximum cell volume initiate rapid division, quickly entering the exponential phase. During the ensuing time points, exponential growth can be observed and the decrease of the forward scatter is a hint for a reduction in mean cell size.

The histograms are plotted from top to bottom during the time course of the experiment
The goal of synthetic biology is to modify biological organisms in an engineering manner using them as nano-factories for a cheap and environmentally friendly production of valuable resources. To reach this goal a detailed genotyping of the corresponding host is a crucial step. Concerning this we sequenced the genome of our host Vibrio natriegens for our synthetic biological purposes.
One of our goals was to identity and characterize chromosomal integration sites. This made the genotyping a critical step for the design of the correct homologous flanks. But the genome map we built became as well a valuable tool for our whole team for the planing and design of genetic constructs.
We got the organism Vibrio natriegens strain ATCC 14048 from the german collection of microorganisms and cell cultures and sequenced it by Illumina sequencing. Illumina sequencing is a next generation sequencing (NGS) method acquiring data by ‘sequencing by synthesis’ (SBS) chemistry. Since 2014 the short read, massively parallel sequencing technique lead to an explosion of sequencing data output and a drop in cost per base pair, launching the next generation of sequencing methods (Illumina). Using Illumina sequencing, we were able to sequence the whole genome in a manner of hours at an affordable cost.
The workflow of Illumina is based on three steps: Library preparation, short read sequencing and genome assembly. For library preparation several kits can be used. Firstly the template DNA needs to be isolated and purified. Then, the DNA fragmentation and tagging are done simultaneously by a method developed by Illumina called tegmentation. Here bead-linked transposomes carrying Illumina sequencing adapters perform DNA fragmentation, normalization and adapter ligation in one step LINK. After that the fragments are amplified and the fragments of suitable size purified. Then, the library can finally be loaded on a flow cell for sequencing. The flow cell is covered by embedded adapter sequences were the fragments can bind to. By bridge amplification the single fragments are amplified into clonal clusters. Finally the clusters are synthesized with fluorescent labeled nucleotides. A designed software records the light’s wavelength and intensity and saves them into a text file containing the different reads LINK. Finally the reads are assembled into bigger scaffolds by bioinformatic software tools like in this case SPAdes 3.12.0 (Bankevich et al., 2012).
We prepared the library using the Nextera DNA Flex Library Prep Kit LINK by Illumina and sequenced the genome with the MiSeq systemLINK with a coverage of 500-fold. The protocols for library preparation and sequencing were given by Illumina LINK. Subsequently we analyzed the data with SPAdes 3.12.0 (Bankevich et al., 2012), Prokka 1.12 (Seemann, 2014) and Geneious 11.1.5.
The Illumina sequencing yielded multiple DNA contigs – short snippets of the genome – that were assembled to bigger scaffolds using SPAdes 3.12.0 (Bankevich et al., 2012). SPAdes is an open source software developed for genome assembly of single-cell data as well as multi-cell data. Version 3.12.0 is available since May, 2018. Subsequently we looked for annotations with Prokka 1.12 (Seemann, 2014). Prokka is an open source software for prokaryotic genome annotations written in Perl. Finally we checked the genome for secondary metabolites using antiSMASH 3.0 (Weber et al., 2015). Here we found three secondary metabolite clusters for nonribosomal peptides (NRPs), bacteriocin and ectoine (see link below for the .gb file).
NRPs are biologically active compounds built by nonribosomal-peptide-synthetases in a ribosome independent manner. These compounds are interesting for clinical applications due to their use as last resort antibiotics, antitumor or antifungal drugs or as immunosuppressants (Walsh, 2008; Strieker et al., 2010). Bacteriocins are heat resistant antimicrobial peptides produced by bacteria to kill or inhibit the growth of other strains without harming the bacteria themselves. Due to their large diversity in structure and function they are interesting for the industry for food preservation or as new potential drugs in the future (Yang et al., 2014). The ectoine cluster may make this organism interesting for further research and for industry applications as well due to the use of ectoine in medicaments against cold and dermatitis or their use in eye drops and nasal sprays (Werkhäuser et al., 2014).
We checked for published genomes on the NCBI database and found two annotations for Vibrio natriegens ATCC 14048. Using Geneious we aligned our sequences against the database and got an 80.5% similarity against the genome with the accession number NZ_CP009977.1 and a 99.97% identity against the genome with the accession number NZ_CP016345.1. Our sequences can be downloaded from the links below.
Both sequences downloaded from the NCBI database have been sequenced with PacBio. The sequence NZ_CP009977.1 has a coverage of 96.48-fold and was assembled with RS HGAP v. 3 while the sequence NZ_CP016345.1 has a coverage of 178.8-fold and was assembled with HGAP v. 2.
Acknowledgements
We would like to thank Dr. Javier Serrania for the possibility and support sequencing the genome of our host Vibrio natriegens. This work became a useful and very valuable tool for our whole team and our whole project and could not have been done without his support.Secondary metabolites
Vibrio natriegens Chromosome 1
Vibrio natriegens Chromosome 2
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology 19: 455–477.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3342519/ (October 18, 2018)
Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069.
https://academic.oup.com/bioinformatics/article/30/14/2068/2390517 (October 18, 2018)
Strieker M, Tanović A, Marahiel MA. 2010. Nonribosomal peptide synthetases: structures and dynamics. Current Opinion in Structural Biology 20: 234–240.
http://www.sciencedirect.com/science/article/pii/S0959440X10000126 (October 18, 2018)
Walsh CT. 2008. The Chemical Versatility of Natural-Product Assembly Lines. Accounts of Chemical Research 41: 4–10.
https://doi.org/10.1021/ar7000414 (October 18, 2018)
Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, et al. 2015. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Research 43: W237–W243.
https://academic.oup.com/nar/article/43/W1/W237/2467910 (October 18, 2018)
Werkhäuser N, Bilstein A, Sonnemann U. 2014. Treatment of Allergic Rhinitis with Ectoine Containing Nasal Spray and Eye Drops in Comparison with Azelastine Containing Nasal Spray and Eye Drops or with Cromoglycic Acid Containing Nasal Spray. Journal of Allergy.
https://www.hindawi.com/journals/ja/2014/176597/ (October 18, 2018)
Yang S-C, Lin C-H, Sung CT, Fang J-Y. 2014. Antibacterial activities of bacteriocins: application in foods and pharmaceuticals. Frontiers in Microbiology 5.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4033612/ (October 18, 2018)
When we first presented our project, many people asked about the mutation rate of Vibrio natriegens, worrying its high growth speed would be accompanied by a higher mutation rate. Some were concerned that random mutations could undo their work in the lab, or lead to a pathogenic Vibrio natriegens mutant, others were hoping this could speed up their mutation experiments.
The mutation rate is the frequency with which new mutations appear in an organism. A mutation in a gene can be a silent mutation, meaning it has no effect, or lead to gene function loss, or alter the genes function. These mutations can be the result of DNA-damage, caused for example by radiation. They can also happen spontaneously through mistakes the DNA-polymerase makes during DNA-replication. The spontaneous mutation rate of most organisms is known, such as Escherichia coli’s and Vibrio cholerae’s. It was determined by selecting for mutants which had changed phenotype, (S. E. Luria and M. Delbrück 1943) or in recent years, as it became cheaper, by analyzing the genome trough genome sequencing. This method is more precise than estimating the mutation rate based on a phenotypical change (Patricia L. Foster 2015) (Marcus M Dillon 2017). The mutation rate plays a major role in multiple arears of biology, influencing for example the rate with which pathogenic microorganisms gain antibiotic resistances or adapt to a new environment (Christopher B Ford 2013).
We conducted mutation experiments to estimate the mutation rate (probability of mutation per cell per division or generation) of Vibrio natriegens by using the number of mutation events and the final number of cells in a culture (Patricia L. Foster 2006). To determine the number of mutation events, mutants have to first be identified. This can be done with rifampicin, an antibiotic which inhibits mRNA transcription by obstructing its elongation path through binding to the β-chain. Some microorganisms can gain a rifampicin resistance through a specific point mutation or deletion in the β-chain of the RNA-Polymerase (Wu and Hilliker, 2017). By plating Vibrio natriegens out on rifampicin plates, thes Vibrio natriegens mutants can be selected.
By testing sister colonies of Vibrio natriegens in this manner and counting the resistant colonies we estimated the mutation rate with the Lea-Coulson median estimator (Lea and Coulson, 1949). For this we inoculated 14 sister cultures of Vibrio natriegens with the same OD600 (About 0.0001) out of an exponential pre-culture. When the sister cultures almost reached the stationary phase, we platted them on rifampicin- and on plats without an antibiotic. After evaluating these plates, we could estimate the Median number of mutants in a culture, by counting all rifampicin resistant cultures and applying the Lea-Coulson median estimator. The final number of cells in the culture was estimated by counting the colony forming units on the plates without antibiotics.
The calculated number of mutations per culture with the Lea-Coulson median estimator was 5,7925 and the estimated final number of cells in the culture were 3165000000 cells. The number of mutations per culture is divided by the final number of cell (Patricia L. Foster 2006). The mutation rate we estimated was 1,83017E-09, and there for a bit lower than the mutation rate of Escherichia coli which was estimated by Luria and Delbrück in a comparable experiment for of about with was calculated to be 3,2E-09 (S. E. Luria and M. Delbrück 1943). Since our number of mutations per culture was estimated to be between 4 and 15 we determined the number of mutations per culture with the Lea-Coulson median estimator, unlike how the mutation rate of Escherichia coli was determined. For Escherichia coli the p0 method was used, because there were between 0,3 and 2,3 mutations per culture (Patricia L. Foster 2006). Because Escherichia coli grows to a lesser OD600, cultures do not undergo as many cell divisions, thus fewer cells have the opportunity to mutate and less mutation events take place. However, our experiment suggests, that the number of mutants per cells are higher in Escherichia coli, meaning that the possibility to choose a mutated colony is higher when working with Escherichia coli.
We are now able to answer the question of our fellow researchers regarding Vibrio natriegens’ mutation rate. After these experiments we know that concerns about unwanted spontaneous mutations when switching from Escherichia coli to Vibrio natriegens are unwarranted. It can be assumed that Vibrio natriegens can speed up mutation experiments anyway, since there are more mutations occurring in the culture, simply because there is a higher density, increasing genetic variability within it. Our data is not conclusive but enables an estimation of the mutation rate of Vibrio natriegens. A more precisely calculated mutation rate can be obtained through whole genome sequencing of multiple Vibrio natriegens cultures as has been done for Escherichia coli (Heewook Lee et al. 2012).
When we first presented our project, many people asked about the mutation rate of Vibrio natriegens, worrying its high growth speed would be accompanied by a higher mutation rate. Some were concerned that random mutations could undo their work in the lab, or lead to a pathogenic Vibrio natriegens mutant, others were hoping this could speed up their mutation experiments.
The mutation rate is the frequency with which new mutations appear in an organism. A mutation in a gene can be a silent mutation, meaning it has no effect, or lead to gene function loss, or alter the genes function. These mutations can be the result of DNA-damage, caused for example by radiation. They can also happen spontaneously through mistakes the DNA-polymerase makes during DNA-replication. The spontaneous mutation rate of most organisms is known, such as Escherichia coli’s and Vibrio cholerae’s. It was determined by selecting for mutants which had changed phenotype, (S. E. Luria and M. Delbrück 1943) or in recent years, as it became cheaper, by analyzing the genome trough genome sequencing. This method is more precise than estimating the mutation rate based on a phenotypical change (Patricia L. Foster 2015) (Marcus M Dillon 2017). The mutation rate plays a major role in multiple arears of biology, influencing for example the rate with which pathogenic microorganisms gain antibiotic resistances or adapt to a new environment (Christopher B Ford 2013).
We conducted mutation experiments to estimate the mutation rate (probability of mutation per cell per division or generation) of Vibrio natriegens by using the number of mutation events and the final number of cells in a culture (Patricia L. Foster 2006). To determine the number of mutation events, mutants have to first be identified. This can be done with rifampicin, an antibiotic which inhibits mRNA transcription by obstructing its elongation path through binding to the β-chain. Some microorganisms can gain a rifampicin resistance through a specific point mutation or deletion in the β-chain of the RNA-Polymerase (Wu and Hilliker, 2017). By plating Vibrio natriegens out on rifampicin plates, thes Vibrio natriegens mutants can be selected.
By testing sister colonies of Vibrio natriegens in this manner and counting the resistant colonies we estimated the mutation rate with the Lea-Coulson median estimator (Lea and Coulson, 1949). For this we inoculated 14 sister cultures of Vibrio natriegens with the same OD600 (About 0.0001) out of an exponential pre-culture. When the sister cultures almost reached the stationary phase, we platted them on rifampicin- and on plats without an antibiotic. After evaluating these plates, we could estimate the Median number of mutants in a culture, by counting all rifampicin resistant cultures and applying the Lea-Coulson median estimator. The final number of cells in the culture was estimated by counting the colony forming units on the plates without antibiotics.
The calculated number of mutations per culture with the Lea-Coulson median estimator was 5,7925 and the estimated final number of cells in the culture were 3165000000 cells. The number of mutations per culture is divided by the final number of cell (Patricia L. Foster 2006). The mutation rate we estimated was 1,83017E-09, and there for a bit lower than the mutation rate of Escherichia coli which was estimated by Luria and Delbrück in a comparable experiment for of about with was calculated to be 3,2E-09 (S. E. Luria and M. Delbrück 1943). Since our number of mutations per culture was estimated to be between 4 and 15 we determined the number of mutations per culture with the Lea-Coulson median estimator, unlike how the mutation rate of Escherichia coli was determined. For Escherichia coli the p0 method was used, because there were between 0,3 and 2,3 mutations per culture (Patricia L. Foster 2006). Because Escherichia coli grows to a lesser OD600, cultures do not undergo as many cell divisions, thus fewer cells have the opportunity to mutate and less mutation events take place. However, our experiment suggests, that the number of mutants per cells are higher in Escherichia coli, meaning that the possibility to choose a mutated colony is higher when working with Escherichia coli.
We are now able to answer the question of our fellow researchers regarding Vibrio natriegens’ mutation rate. After these experiments we know that concerns about unwanted spontaneous mutations when switching from Escherichia coli to Vibrio natriegens are unwarranted. It can be assumed that Vibrio natriegens can speed up mutation experiments anyway, since there are more mutations occurring in the culture, simply because there is a higher density, increasing genetic variability within it. Our data is not conclusive but enables an estimation of the mutation rate of Vibrio natriegens. A more precisely calculated mutation rate can be obtained through whole genome sequencing of multiple Vibrio natriegens cultures as has been done for Escherichia coli (Heewook Lee et al. 2012).
After creation of the Marburg Collection, we wanted to characterize the parts in V. natriegens. When we started with our project, we had no clue about the behavior of the genetic parts that were integrated into our toolbox. Previous research mainly focused on microbiological description rather than characterization of synthetic constructs as we already discussed in our V. natriegens review (Link to Description).
We decided to characterize the parts in our Marburg Collection and hence we did pioneering work to provide the scientific community the data that enable rational utilization of V. natriegens for various applications in synthetic biology.
Before acquiring the final data, we established a fast and convenient platereader workflow that is tailored to the fast growth rate of V. natriegens. We demonstrate its superior performance and discuss considerations in terms of plasmidal context and data analysis on our measurement page (Link to Measurement).
After having established an experimental and data analysis workflow and after determining the optimal plasmidal context for reporter experiments, we started to apply our knowledge to characterize the parts in our Marburg Collection.

Data were normalized over the strongest construct J23100. Error bars represent the standard deviation of the measurements of three independent experiments
Characterization of pTet
In addition to constitutive promoters, the Marburg Collection contains two inducible promoters, pTet and pTrc. For all experiments with inducible promoters, we added the respective inducer concentration to the preculture as well as to the main culture to ensure constant expression.The first experiments were performed with the pTet promoter that can be induced by the tetracycline derivative anhydrotetracycline (ATc). ATc is much less cytotoxic but still capable of binding and altering the structure of the repressor TetR, leading to release of the promoter and enabling transcription.To measure the dose response behavior of the pTet, we made a dilution series of ATc. Following the recommendation of our advisors (Stefano Vecchione), we started with the concentration commonly used in E. coli, started with the concentration (100 ng/mL). The starting concentration was diluted twofold in 20 subsequent steps. Our results are shown in figure 2. The absence of bars for the four highest concentrations is due to the fact that the cultures did not reach an OD600 of 0.2 in the six hours of the measurement.
Remarkably, we observed reasonable growth of those same cultures in the preculture already induced with the identical amount of ATc. Knowing that luminescence is produced at the end of an enzymatic cascade, starting with intermediates of the phospholipid metabolism ( Meighen 1991), we reckon that very strong induction could decrease the fitness of cells and that after dilution in room temperature medium, strained cells are not able to recover from the stationary phase.
However, we only observed this phenomenon in experiments with pTet, although we obtained higher signals for the strongest constitutive promoters as well as for the highly induced pTrc. We checked for toxicity of ATc but could not see a measurable effect. Another possibility is that TetR interacts with components inside the cell and that high ATc increases these interactions.
Blast searches of TetR against the genome of V. natriegens identified one protein that shares some homology with the N-terminal part of TetR which could result in cross talk between the host and the inducible promoter.

J23100 was used as positive control and for normalization. Error bars represent the standard deviation of the measurements of three independent experiments
pTet allows relatively tight control of gene expression and is therefore well suited for driving the expression of potentially toxic proteins. On the other hand, we were not able to induce strong expression that can compete with strong constitutive promoters or the fully induced pTrc.
Characterization of pTrc
pTrc is the second tested inducible promoter. It contains lac operator sites and is therefore regulated by the repressor LacI which is constitutively expressed from a downstream gene.pTrc can be induced Isoopropyl-β-D-thiogalactopyranosid (IPTG), a chemical derivative of lactose ( Camsund et al.2014). Similar to our experiments with pTet, we made a dilution series starting with the commonly used IPTG concentration for E. coli 0.5 mM. We observed a five fold induction and a saturation that occurred at a dilution of 2^5 (~15 µM). The strongest expression is similar to the expression gained from the strongest constitutive promoter J23100 while the expression in the absence of inducer equals medium strong promoters.
As a consequence, we do not recommend using pTrc in constructs where a tight control of gene expression is desired. Instead, pTrc is well suited when strong expression is required.

J23100 was used as positive control and for normalization. Error bars represent the standard deviation of the measurements of three independent experiments
Taking the results of both inducible promoters into account, we made two observation. In both cases, the dynamic range is smaller compared to E. coli and the inducer concentration that facilitates saturation is 32 and 64 fold lower for pTrc and pTet, respectively, than the concentration that is typically used for E. coli. A possible explanation could be found in the fast growth of V. natriegens which might result in a lower concentration of the repressor proteins in the cells, finally leading to a less restricted control of the negatively regulated promoters. However, we do not have experimental support for our idea.
One novel key feature of our toolbox are the connectors. They were designed in order to function as insulators to prevent crosstalk between neighboring transcription units (Link to Design). Therefore a perfectly insulating connector would prevent the readthrough from backbone sequences that most probably caused the notably high expression that was measured in the promoter experiment for the dummy promoter. In addition to blocking transcriptional readthrough, a good connector must not possess any cryptic promoter activity.
We focused on characterizing the 5’ Connector because we expect the stronger influence on signal strengths. For characterizing our connector parts, we created 20 test plasmids with the lux operon as the reporter.
In our toolbox we provide five short connectors, which solely possess the fusion sites for LVL2 cloning, and five long connectors which additionally harbor self-designed insulators (Link to Design).
Each of these ten connectors were cloned with the constitutive promoter J23100, to check for effects on an active promoter, and with the Promoter Dummy to quantify the extent of transcriptional activity that reaches the Promoter Dummy.
 A
A B
BResults of Connector measurments
A) Connector constructs built with J23100 as promoter part
B) Connector constructs built with the Dummy Promoter as promoter part
For the constructs built with small connectors, we also observed varying signals but to a lesser extent compared to the long connectors (figure 4, B).
For all ten connectors that are provided in our toolbox, we show a tenfold range in the measured luminescence/OD600 signal. As a conclusion, we recommend to carefully consider the combination of promoter and 5’ Connector for rationally designing constructs.
Taking a look at the constructs that were built with the Promoter Dummy, we also see a huge difference in the expression signals. For the long connectors we expected a negligibly low reporter expression which we observed for two out of five long 5’ Connectors resulting in a 14 fold signal reduction compared to the “Promoter Dummy” reference. We interpret this result as the confirmation that those two connectors function as insluators!
The remarkably strong signal observed for the remaining three connectors could be due to inefficient terminators or cryptic promoters in the pretended “neutral sequence”.
For the remaining five constructs possessing the five short 5’ connectors we observed a range from 0.3 to 5.5 fold compared to the “Promoter Dummy” reference. We are not able to give an experimental explanation for this observation but we could imagine that the LVL2 fusion sites, the only four bases that differ in these constructs, could constitute a weak promoter together with surrounding sequences.
Summarizing the connector characterization, we found that sequences upstream of short synthetic promoters greatly affect reporter expression, which is in accordance with literature ( Carr et al.2017). Moreover, we demonstrated that two of our five self-designed connectors efficiently reduce the signal resulting from other sources than the actual promoter. We additionally conclude that algorithms that predict the “neutrality” of sequences alone are not sufficient to create well functioning insulators.

LVL2 plasmids were created for these experiments consisting of a RFP transcription unit with the strong constitutive promoter J23100, followed by the lux operon with the promoter dummy. The terminator located at the 3' end of the RFP transcription unit is the part which is characterized in this experiment.
With this setup, transcriptional activity of the RFP reporter is blocked by the terminator. Therefore the measured luminescence signal can be seen as an indicator for the efficiency of the terminator.
As discussed previously, RFP is not suitable for precise quantitative characterizations (Link to Measurement). Therefore we did not calculate ratios of the reporter upstream and downstream of the tested reporter as was described in previous experiments ( Chen et al.2013). However, we used the existence of an RFP signal as control for the correctly assembled test constructs and for the activity of the promoter driving RFP.

Displayed is the relative luminescence signal to the control construct J23100 obtained for constructs with the respective terminator. Error bars represent the standard deviation of the measurements of three independent experiments.
The data shown in figure 6 were acquired and analyzed following our novel workflow described on the measurement page (Link to Measurement). Like in all previous experiments, the obtained raw data for each sample were normalized over the construct J23100 from the promoter characterization.The strongest signal was observed for B1002 and B0010 with a relative signal 0.65 and 0.50 respectively, suggesting these two terminators as rather inefficient. In contrast, we could show a signal reduction four and eight fold for B0015 and B1006, respectively.
By comparing our data to the characterization provided in the iGEM registry for E. coli, we can show that the general trend is similar for both organisms. Exemplary, the terminator with the highest described efficiency in E. coli (B1006) also was found to reduce the luminescence signal most in our experiments.
In general, we found stronger signals for the reporter downstream of the terminator than what was described for E. coli. However, it has to be noted, that we used the highly sensitive reporter Lux instead of a fluorescent protein. Therefore we assume that we were able to detect a higher degree of transcriptional readthrough, which would not be distinguishable from the background when using a fluorescence reporter.
However, we cannot exclude species specific differences that cause a generally higher degree of transcriptional readthrough over terminators for V. natriegens compared to E. coli
In addition to the terminators shown in figure 6 we also measured a test construct possessing B1003. The, by far, lowest signal was found for B1003 with a 15000 fold reduction of the signal. To ensure that this extremely weak signal is authentic and not caused by cross talk from neighbouring wells, we tested this sample in an otherwise empty 96 well plate and confirmed the general existence of a signal. However, we can not exclude the presence of single mutations within the lux operon dramatically diminishing the overall generation of luminescence. This result was not displayed in figure 6 to omit extreme stretching of the Y-Axis thus loosing visual information for the other tested terminators.
For quantifying the RBS strengths, unfortunately, we could not use our favorite reporter the lux operon because in case of this operon, each CDS possesses its own RBS. Therefore, replacing the RBS upstream of the first CDS (LuxA) alone does not suffice to achieve a difference in reporter expression dependent on the RBS strength. Consequently we used sfGFP, the reporter that showed the second best performance in our initial reporter experiment (Link to Measurement).

Test constructs were built with the same parts except for the RBS part. The sample "Empty" represents V. natriegens with a plasmid without a sfGFP reporter. The error bars indicate the standard deviation of four technical replicates.
We expected that the order of the strength of the tested RBS should be similar to E. coli. Prior to the experiment, we created an alignment of the 16S rRNA of both organisms and found that the Anti-Shine-Dalgarno sequence, the bases that are responsible for binding the Shine-Dalgarno sequence on the mRNA, do not differ between both organisms (figure 8).

The Anti-Shine-Dalgarno sequences is indicated with an additional annotation. The 3' sequence of the 16S rRNA does not differ between both organism
On the measurement page (Link to Measurement). we suggested the lux operon as the reporter of choice for all experiments that focus on measuring transcriptional activity . We see the RBS characterization as the confirmation that using sfGFP as reporter does not yield reliable data for very weak expression.
In future experiments, enzymatic reporters such as LacZ or β-glucuronidase (GUS) could be tested for their suitability in experiments for the quantification of translational efficiency or post translational effects (e.g. degradation tags)
Our toolbox contains the three degradation tags M0050, M0051 and I11012. Similar to our RBS experiments, we could not use the lux operon as a reporter because the degradation tag is only added to the C-terminal end of the last enzyme encoded in the operon. Therefore, we again used sfGFP as reporter and test constructs were designed with one of the tags fused to the CDS of sfGFP. A non-tagged sfGFP construct serves as the reference.

The respective degradation tags were appended to the C-terminus of sfGFP. The error bars indicate the standard deviation of four technical replicates.
As expected, appending a degradation tag to a protein decreases its concentration. The strongest decrease and therefore the highest degradation was shown for M0050 with a signal that is not distinguishable from the non-sfGFP expressing control. The second strongest signal reduction was shown for I11012, followed by M0051, which showed the least efficient degradation (figure 9). Our results are in qualitative accordance with the description of these tags for E. coli, which are provided in the registry.
The tested degradation tags belong to the family of SsrA degradation tags. Naturally, they help to degrade incompletely translated proteins by labeling them for Clp proteases (Farrell et al. 2015). We checked for the presence of ClpP, one of the proteases involved in degrading SsrA tagged proteins in E. coli and found a highly homologous protein in V. natriegens. Therefore we assume that the mechanism of Clp mediated degradation is conserved between both organisms, which is in accordance with our results.
Each organism has a preferred codon usage that affects the efficiency of translation through the abundance of tRNAs (Rocha, 2004). The tRNA composition differs between organism which can result in a loss of protein expression. The goal of our project is to replace E. coli in as many applications as possible. We know that scientists all over the world have been extensively working with E. coli for decades and have collected huge collections of plasmids for this organism. We already showed in many experiments that parts taken from E. coli are in general functional in V. natriegens.

The error bars indicate the standard deviation of four technical replicates.
Origins of replication (Oris) are genetic elements where DNA replication is initiated. In plasmids the Ori sequence is responsible for its maintenance and for the copy number inside the cell (Selzer et al., 1983; Brantl, 2014).
The origins of replication colE1, pMB1 and p15A belong to the same family. They do not code for any enzyme but are replicated by the hosts RNA polymerase (Cesareni et al., 1991; Brantl, 2014). The polymerase transcribes a region 508 bp upstream the Ori sequence (Tomizawa & Itoh, 1981; Selzer et al., 1983) synthesizing a pre-primer RNA called RNA II. During transcription the RNA II underlies conformation changes building secondary structures(Brantl, 2014). This structures contain typical loops (Cesareni et al., 1991) that binds to the plasmids’ Ori sequence building an RNA-DNA hybrid (Cesareni et al., 1991; Brantl, 2014). The RNA II is than cleaved by the hosts RNase H to become a mature primer (Cesareni et al., 1991; Brantl, 2014).
Results
For our collection we characterized three Oris commonly used in molecular biology: colE1, pMB1 and p15A. We measured two different plasmids, one with and another without a LUX cassette. Both plasmids consist of a kanamycin resistance cassette and one of the three Oris described (colE1, pMB1, p15A). The LUX expression plasmid contained a constitutively expressed LUX cassette of ~6kb (figure 1). The other one contained a connector sequence to build an ‘empty’ plasmid. By comparing this constructs you may consider that the copy number is not only influenced by the LUX expression but also by the plasmids sizes. This Oris belong to the same family differing in mutations in the RNA I region (Tomizawa & Itoh, 1981; Selzer et al., 1983).
We measured the plasmids’ copy number by qPCR using the absolute quantification method.
A qPCR is set up the same way like a normal PCR but with addition of a DNA binding fluorophore in this case SYBR Green. SYBR Green binds double stranded DNA emitting a high signal while unbound SYBR Green shows only low fluorescence (Zipper et al., 2004). In every PCR cycle the number of double stranded DNA is duplicated emitting an increasing fluorescence signal. This signal is detected after every cycle by the qPCR machine and the value is saved. After the run finished, normally after ~40 cycles, a signal threshold is determined and the corresponding cycle when the threshold was reached is saved for further analysis.
For the qPCR run first total DNA from our host containing the plasmids of interest was isolated in the exponential phase (OD600 ~ 0.5), purified using the innuPREP Bacteria DNA Kit from Analytik Jena and all samples normalized to ~5ng/ul with the Qubit 2.0 fluorometer from ThermoFisher scientific. Subsequently a dilution series was made in 1.5ml tubes diluting the DNA 7 times 1:2. This way the dilution series contained 8 steps reaching from 20 to 2-7. Two different primer pairs were used for the analysis: one matching the housekeeping gene dxs present once on the genome and the other matching the kanamycin resistance cassette on the plasmid. We choose the dxs sequence to be able to compare our results to the values reported by literature for E. coli. The DNA samples used for the amplification of the kanamycin cassette were the same used for the dilutions 2-4 and 2-5. The threshold cycles (Ct) acquired in triplicates from the dxs sequence were used for a standard curve. By comparing the Ct values from the resistance cassette with the corresponding standard curve the number of copies could be determined as multiples from the dxs sequence. It should be considered that the dxs sequence is coded on the first chromosome of V. natriegens at ~ one o’clock. Due to that probably the sequence is present more than once because of multifork replication of the genome.
To build the standard curve the Ct values were plotted on the y-axis and the corresponding dilution steps on the x-axis. The x-axis was set logarithmic and the standard curve was calculated with Excel. The curve’s formula was then used to calculate the corresponding x-value from the resistance cassette’s Ct values. Because the x-values describe a theoretical dilution the Ct values were multiplied with this value and with their corresponding dilution to obtain the final amount of multiplies compared to the genome. For every Ori an own standard curve was calculated.
In our experiments we showed that the plasmids’ copy number controlled by three different Oris differ a lot when comparing V. natriegens with E. coli.
One possible explanation might be different expression levels of RNA I and RNA II respecting the rate of RNA I – RNA II bounds (Cesareni et al., 1991) due to the divergent metabolism in V. natriegens and E. coli. Another plausible explanation might be the different methylation patterns in both organisms probably affecting the formation of the RNA II secondary structures and subsequently its binding affinity to the DNA (Russell & Zinder, 1987; Cesareni et al., 1991).
It was shown that mutations especially in the loop I structure might be responsible for Ori compatibility and copy number control (Selzer et al., 1983; Cesareni et al., 1991). The copy number is mainly determined by two factors: the binding efficiency of the RNA II to the DNA – specially controlled by the stabilization of stem-loop IV – (Cesareni et al., 1991) and the interference of the complementary RNA I to the RNA II pre-primer (Brantl, 2014). The RNA I is transcribed constitutively from the complementary strand from RNA II pre-primer (Brantl, 2014). Binding of RNA I to RNA II prevents the correct folding of the pre-primer (Brantl, 2014). This way the RNA-DNA hybrid can not be formed and subsequently the primer maturation can not take place (Brantl, 2014).

Acknowledgements
We would like to thank Marcel Wagner for his support during our project with the quantification of plasmids copy number, the set up of this experiments and the handling with the qTower. Thanks to his support we were able to perform and evaluate this experiments to fulfill this study.Natural Competence
 In our project we wanted to build many different strains. For this we have to make many genomic changes of the wildtype of Vibrio natriegens. Natural transformation is a good method for multiplex genome editing in a short time (Figure 1).
In our project we wanted to build many different strains. For this we have to make many genomic changes of the wildtype of Vibrio natriegens. Natural transformation is a good method for multiplex genome editing in a short time (Figure 1).



Lvl0
For implementing and domesticating the tfox gene into our Marburg collection, we had to built a Level 0 plasmid. At first we amplified the tfoX gene from the pMMB-tfox plasmid (Daliaet al.2017) and build a Lvl 0 plasmid to integrate this part in the Marburg Toolbox (if you want to see how to build a level 0 plasmid take a look at the Marburg Collection). After we were able to construct and build the plasmid, we first did a test digest with EcoRI and PstI to see if it was correct (Figure 4). Then we sent the plasmid for sequencing, we also received confirmation of the accuracy of our plasmid.

Lvl1
In order to construct two plasmids with full transcription units, with two different promoters we assembled tfoX together with the constitutive promoters J23100 or J23110 into a transcriptional unit by golden gate assembly.
We suggest that these problems could be caused because tfox is maybe somehow toxic to E. coli cells.

tet promoter
In addition to constitutive promoters, the Marburg Collection contains two inducible promoters, pTet and pTrc. To build our level 1 plasmid we chose the pTet promoter. The pTet promoter can be induced by the tetracycline derivative anhydrotetracycline (ATc). ATc is much less cytotoxic than IPTG but still capable of binding and altering the structure of the repressor TetR, leading to release of the promoter and enabling transcription.
Establishing Flp/frt in V. natriegens
One of our goals was to create fast genomic modifications, establishing the Flp/frt system in V. natriegens. Therefore, we assembled a plasmid encoding the FLP recombinase, amplified from pBR-flp into the backbone K2560002, creating a Lvl 0 plasmid by Golden Gate assembly. Next, we used this created part K2560271 to assemble a Level 1 plasmid, encoding a full transcriptional unit under the control of a constitutive promoter. This plasmid was transformed into a V. natriegens strain with a successfully integrated chloramphenicol resistance cassette by electroporation. The resulting clones were tested by colony PCR and positive clones were cultivated in LBV2-Kanamycin. The three-phase streaking pattern on LBV2-Kanamycin was performed to get single colonies. These colonies were streaked on replica plates with LBV2-Kanamycin and LBV2-Kanamycin-Chloramphenicol to screen for chloramphenicol sensitive clones. Of 100 clones, only eleven clones were not chloramphenicol sensitive. Eight chloramphenicol sensitive clones were tested by four different colony PCRs: one to amplify the region of the gene of interest, a second PCR to amplify the upstream and chloramphenicol fragment, a third PCR would amplify the downstream-chloramphenicol fragment and the last PCR would amplify only the chloramphenicol cassette. The first PCR should result in a fragment size of 2400 bp in case of a successful excision mediated by the FLP recombinase. In the wildtype genotype, this PCR would result in a fragment size of 3900 bp. The second, third and fourth PCR would not show a fragment in case of the chloramphenicol excision.
In all eight tested clones, the excision of the chloramphenicol resistance cassette was proved.


Here we just show that Flp mediated excision is working. In future the functionality of Flp mediated insertion, translocation and inversion need to be demonstrated.



VibriClone
To build our first VibriClone we investigated which deletion is the most important one for a cloning strain. It has been shown for V. cholera that the transformation efficiency greatly increases when the two extracellular nucleases are knocked out. Thus, the DNA to be transformed into the organism is no longer degraded (Blokeschet al.2008) . In order to increase our transformation efficiency, we decided to delete the dns as our first target as it has been reported, that a knockout improves the plasmid amount and quality. Furthermore, for transformation by chemical competent cells or natural transformation the deletion is one of the crucial parameters.The first step of creating a Δdns deletion strain was to build a selected product with the upstream and downstream regions of the dns (~500bp) and the antibiotic cassette chloramphenicol (~940bp). frt sequences, needed for deletion of CmR are used as overlapping sequences. The same amount of every fragment was used for the PCR. We were able to successfully create the fragment (Figure 1 A). V. natriegens is able to take linear DNA Fragments up and integrate it by homologous integration into the genome. With the linear DNA fragment, it was possible to delete the dnsV. natriegens strain without genetic modifications. To check if the deletion was successfully we made colony-PCRs with primers binding inside the inserted fragments (1. upstream-CmR -downstream: ~1940bp; 2. CmR -downstream: ~1440bp; 3. upstream-CmR: ~1440bp). We could prove that the deletion cassette was inserted into the genome (Figure 1 B).
The next step was to cure the antibiotics cassette. In order to leave the possibility open to work with plasmids with a chloramphenicol resistance, it is very important to delete the inserted antibiotic cassette. Because of the frt-sites around the CmR it is possible to to cut it out. Therefore we use the pBR-Flp plasmid and transform it into the cells. Introducing this plasmid into V. natriegens and expressing Flp, the recombinase will recognize the integrated frt sites. By Flp, mediated recombination the resistance cassette will be cut out of the genome, creating the deletion of the desired gene.
A colony PCR, negative for CmR will prove the loss of resistence and the strain is almost ready for the usage. Therefore we used primers binding inside and outside of the cassette (1. upstream-CmR -downstream: ~1940bp; 2. CmR: ~940bp; 3. CmR -downstream: ~1440bp; 4. upstream-CmR: ~1440bp). We could prove that the CmR was cut out of the genome (Figure 2).


We were able to design and build the first VibriClone strain - VibriClone 1.0. With this achievement we can give the scientific society a possibility to work faster, get a higher efficient in transformation and to get faster results. However, as the name VibriClone 1.0 suggests it gets even better. Therefore we started with an upgrade called VibriClone 2.0.
VibriClone 2.0
The upgrade of the cloning strain should even have more practical and user-friendly features as the first one has already. We would like to knock out both exonucleases (Dns and ExeM) so that it would be possible to transform DNA without degrading. In addition, we would like to insert a point mutation in recA, that the homologous recombination between plasmids and into the genome. Moreover, we also want to make it possible to store in the fridge. Therefore we insert the katG gene encodes an enzyme with an catalase and peroxidase activity into the genome. Furthermore, we also wanted to make the blue-and-white selection possible for our VibriClone, we can achieve that by integrating the lac operon into the genome.Exonuclease
As selected product we use the dns cassette. To use the cotransformation we build a cassette with 3000bp homologous regions upstream and downstream of the dns and the chloramphenicol cassette in the middle. It was possible for us to amplify the individual fragments and in each case to fuse the upstream or the downstream fragment to the chloramphenicol cassette. Unfortunately we were not able to build the whole fragment yet.

RecA
For our VibriClone 2.0, we would like to have a modification in the gene recA. It should be a point mutation from G to A at position 720. Thereby the glycin at the place 158 chance to aspartic acid. We use this model to modify our strain VibriClone. To insert a point mutation in recA, we built two fragments with the mutation in the overhang. It was possible for us to use a fusion PCR to construct an unselected product that contains the point mutation. To check if the point mutation really took place, we sequenced the PCR product. The result clearly showed the modification we wanted to achieve.
KatG
Another feature, which would improve the applicability of V. natriegens as a cloning chassis, would be to engineer cold resistance, due to fact that we all are used to store our plates of E. coli in the fridge. For integrating the katG into the genome, we combined introducing katG with the deletion of the second nuclease by utilizing 3000 bp homologous overhangs of the upstream and downstream flanking genomic region of exeM. We were able to build this unselected fragment successfully (exeM upsteam - katG - exeM downstream: 8327 bp) (Figure 6). With this unselected product it is possible to perform a co-transformation and to integrate the point mutation into the genome.
The lac operon
In order to use the 𝛼-complementation we build a cassette consist of the upstream and downstream regions of lacZ and the lactose operon from DH5𝛼. Thus, it is possible to introduce the lactose operon by cotransformation as an unselected product with the other described products.The lactose operon of E. coli has a size of 6326bp. Since that is very large we decided to use smaller homologous flanks. It was shown that the natural co-transformation work well with 1000bp flanks, so we use this size. Otherwise the linear DNA Fragment has a size of ~12000bp. In this case it was a good decision because to get the smaller fragment with 8342bp was hard enough. It only worked once to fuse the three fragments. But then the result could not be reproduced.
Cotransformation
In 2017 the group of Ankur B. Dalia showed that it was possible to integrate linear PCR products with an selected product containing an antibiotic cassette that replaces a gene and unselected products to modify the genome in the way you like in V. natriegens by using natural transformation (Daliaet al.2017) . Our plan was to use this method to perform the genetic changes that we have considered and where we have built the fragments for. Since we were not able to build the dns deletion fragment with 3000bp, we built a smaller cassette (1000bp) in parallel to start creating the VibriClone 2.0 by using natural transformation. So far the natural transformation with the small selected product and the unselected products of the genomic modifications don´t work. This is not surprising because it was shown that the efficiency is much lower with 1000bp flanks (Daliaet al.2017) . We wanted to try it nevertheless. However it is very important to get the selected product with big homologous regions. Thus it would be possible with a high efficiency to carry out the modifications and obtain our improved strain - VibriClone 2.0.

Those three fragments were transformed into V. natriegens by natural transformation. The transformation in general worked and we got clones in which the dns deletion cassette integrated. This was proved by colony PCR. In theory, the T7 integration cassette and the lon deletion cassette should also integrate to the V. natriegens genome at the same time. However, co-transformation of these linear fragments did not work because of more than 200-screened clones no one was showing the desired genetic modifications of lon deletion or T7 integration.

This could be due to the big fragment sizes of the lon and T7 cassettes (from 6000 – 8000 bp). Another reason why the co-transformation did not work could be that it is recommended to use a selected product with long up- and downstream homologous regions. Up to a size of 3000 bp the co-transformation efficiency increases with the size of the homologous region (Hayes et al. 2017) . We used fragments with 1000 bp up- and downstream- homology because we were never able to fuse the 3000 bp up and downstream fragments to the chloramphenicol resistance cassette but still tried to perform co-transformation although there was a small chance to get a clone with the integration of the desired modifications. For future work, fragments that are difficult to fuse to each other could be created in a plasmid based manner and introduced into the V. natriegens cells by conjugation (Hesek et al. 2016) .
Another promising solution for the integration of the T7 system is the utilization of the Flp/frt system for Flp mediated insertion. We could already demonstrate, that genomic excisions by FLP recombinase are possible in V. natriegens. The next step would be to amplify the T7 cassette with FRT site overhangs and to use this fragment for integration at the locus where dns was deleted by FLP mediated excision of the chloramphenicol resistance cassette, leaving a frt site behind. Unfortunately, this could not be tested due to the lack of time.
Nevertheless, we were able to show, that V. natriegens is suitable for the production of proteins because we could successfully overexpress and purify proteins using tcr and T5 promoter based systems. See results of the Metabolic Engineering sub-project.
V. natriegens is a suited chassis to test protein interactions based on B2H
The first step, after the theoretical work for establishing VibriInteract, was to check if V. natriegens ATCC 14048 is suitable for use in the conventional B2H system. For this, we plated wildtype cells on different agar plates. On LBv2 agar plates supplemented with 0.5 mM IPTG and 40 µg/ml Xgal, we could observe growth but no blue colour of colonies. This led us to thinking that the lac operon is probably not functional or that the β-galactosidase encoded by lacZ, which is needed for blue white screening is not functional. Later on, we confirmed that lacZ of V. natriegens lacks amino acids in the midsection. Hereby, we learned that we can’t use blue-white screening for our interaction studies. On M63 minimal medium supplemented with maltose we could observe growth after less than a day. Thus, we learned that our wildtype strain is able to utilize maltose. We got the same result when plating V. natriegens in MacConkey agar supplemented with maltose. On those plates we could observe a red colour of colonies which means that acidification takes place which led us to thinking that cells utilized maltose with acids as byproduct. This means that V. natriegens is a suited chassis to test protein interactions based on B2H.

The adenylate cyclase of V. natriegens was deleted successfully
Next, we established a ΔcyaA deletion strain. Therefore, we created insertion cassettes as described. Learn more about the design of our deletion cassettes following this link! V. natriegens could successfully be transformed with the deletion cassette using natural transformation. Learn more about natural transformation following this link! We did test PCRs to verify our integration using primers inside and outside of the cassette. Primers outside of the cassette should show shorter fragments in our integration clone compared to wildtype (cyA::CmR: ~2300bp; wildtype: 3900bp). Primers inside of the fragment should only show fragments if the cassette was integrated into the genome (CmR: ~940bp; cyA-upstream-CmR: ~1600bp; CmR-cyA-downstream: ~1600bp). When checked on an agarose gel, we could verify that our deletion cassette was inserted into the genome of V. natriegens (Figure 2).
Then, we had to loop out the resistance cassette. The correct clones were transformed with a level 1 plasmid designed by ourselves carrying the gene encoding the Flp recombinase which recognizes the frt site. Transformation clones were tested via test PCR to examine if they carry the flp recombinase (not shown here). Correct clones were cultivated and streaked onto plates. Of those plates, single colonies were restreaked onto replica plates supplemented with and without chloramphenicol. 11 out of 100 clones in total showed sensitivity for chloramphenicol. Clones only growing on the plate without chloramphenicol were tested via test PCR. For this purpose, we used the same primers used for verification of insertion clones. Wildtype and our insertion mutant should show the same fragment sizes mentioned above. If the second homologous recombination was successful and the clone no longer carries a chloramphenicol resistance, only primers outside of the deletion cassette should result in a fragment (ΔcyA: ~1440 bp) (Figure 3). We could successfully get rid of the chloramphenicol resistance in every clone tested. With this, we were a step closer to establishing VibriInteract.

VibriInteract can be used for complementation in protein interaction studies based on B2H
After establishing VibriInteract, we confirmed that it is no longer able to utilize maltose as a carbon source. We plated VibriInteract (ΔcyA) together with wildtype as a control onto MacConkey agar plates supplemented with maltose. We expected VibriInteract to show white colonies, since it should no longer be able to utilize maltose. Unfortunately, we observed red colonies on MacConkey plates (Figure 5, top). This suggests that some sort of acidification occured during the fermentation of a substrate. V. natriegens was shown to be able to form acids by mixed acid fermentation and to especially form formate and acetate under aerobic conditions. Under anaerobic conditions, cultures form mainly formate. (Hoffart et. al, 2017) . There are three substrates present in the medium that could be responsible for the outcome of this experiment. The first probable substrate is lactose which is an ingredient of the ready made MacConkey agar. Although we hypothesized,based on bioinformatic analysis and its missing ability to cleave Xgal, that the lac operon is not functional in V. natriegens, it could still be working. Maybe the β-galactosidase cannot cleave Xgal but could still degrade lactose. This could be tested by preparing MacConkey agar without lactose to check if colonies still turn red. The second feasible substrate is peptone which consists of peptides and aminoacids. The last one is maltose, which could still be somehow degraded by V. natriegens. This is not likely to have occured because fortunately, we also plated VibriInteract together with wildtype as a control onto M63 minimal medium supplemented with maltose. On those plates, we could observe growth for wildtype but nearly no growth for VibriInteract (Figure 5, bottom). Our deletion was successful and the strain can no longer utilize maltose successfully. There was still some growth of the strain on minimal medium because absence of adenylate cyclase and consequently absence of cAMP does not lead to completely abolished expression of the regulated genes but to a 50-fold reduction in gene expression. Thus, we conclude that we can use M63 minimal medium supplemented with maltose to test for complementation of adenylate cyclase to perform protein interaction studies based on B2H. By this, conditions for the first test of a Vibrio Two Hybrid (V2H) were all set.

Plasmids used for B2H assay are applicable in V2H
We then certified that pUT18 and pKT25 plasmids used in B2H assays also work in V. natriegens. For this purpose, we transformed VibriInteract with plasmids of the commercially sold Euromedex B2H kit. As negative control, cotransformation of cells with the bait plasmids pKT25 and pUT18C were performed. As positive control, cotransformation of cells with pKT25-Zip and pUT18C-Zip plasmids were performed. Transformation clones were restreaked and later on inoculated in LBv2 supplemented with corresponding antibiotics. Those cultures were spotted onto M63 minimal medium supplemented with maltose. Eight clones of each transformation were tested. Six out of eight clones containing negative control plasmids showed little growth while two clones did not show any growth (Figure 6, left). Seven out of eight clones containing positive control plasmids showed normal growth while one clone showed very little growth (Figure 6, right). In general, we see a clear difference between the positive and the negative control. The positive control has an increased growth compared to the negative control. Consequently, we can use plasmids of the B2H assay in our V2H assay. Amazingly, spotted cultures could be analyzed after only 6 hours of incubation at 37 °C. For B2H assays in the E. coli strain BTH101 it is recommended to incubate plates for four to even eight days!
VibriInteract grows faster than BTH101
For further characterization we did a growth experiment to compare the doubling time of VibriInteract to the doubling time of our wildtype strain and to the doubling time of BTH101, the E. coli strain commonly used for B2H assays. Therefore, we diluted overnight night cultures of the three strains to an ΔOD600nm of 0.05 in LBv2 medium for V. natriegens strains or in LB medium for BTH101. The baffled flasks were incubated shaking at 37 °C. In general, we could observe that VibriInteract has a slighty slower growth rate with a doubling time of approximately 18 min compared to the doubling time of approximately 16 min of our wildtype strain in this assay. But when comparing the doubling time of approximately 18 min to the doubling time of the E. coli strain BTH101 of approximately 25 min, we could verify that VibriInteract still has a faster growth rate. VibriInteract did not reach an OD similar to wildtype in this experiment. This could be due to the low level expression of catabolic enzymes of mainly fermenting processes which are regulated by cAMP. The final OD would’ve been important for VibriExpress but for VibriInteract the density of a culture only plays a minor role since it is not a producer strain. To conclude, VibriInteract has a higher growth rate than BTH101 which is the commonly used strain for Bacterial Two-Hybrid assays.
VibriInteract shows modified cell morphology
We could observe a slower growth rate of VibriInteract in comparison to wildtype. Thus, we decided to analyze cells concerning their cell morphology since defects in growth can often be due to defects general cell growth, inefficient cell division etc which can be examined using microscopy. For this purpose microscopic pictures of VibriInteract cells and wildtype cells were taken. Interestingly, what we could observe are what looks like chains of cells next to normal looking single cells. Those chains weren’t made of single cells but rather cells that could not complete cell division completely. In our analysis we could also observe such cells for wildtype but in a much lower number. In earlier experiments, we discovered that cells start to expand upon provision of fresh medium but without undergoing cell division using flow cytometry (See Part Collection results). In this experiment these chains of cells would have been defined as one event instead of several. In contrast to the mentioned experiment, cells of VibriInteract imagined here were taken from exponentially growing cultures instead of freshly inoculated cultures. cAMP is not only a key regulator of many catabolic processes but is also involved other major regulatory processes. One these processes is cell division regulation. It was suggested before that cAMP plays a role in regulating a mediator protein between PBP2 and the separation apparatus in E. coli (D'Ari et al., 1988) . V. natriegens cells could also be affected in a similar manner. This would explain the phenotype of VibriInteract. Although cells show this abnormal phenotype, plasmids of the B2H assay were functional in VibriInteract and cultures exhibited a sufficient growth rate.
In our project, we provided a first indication of a successful mutation via CRISPR/Cas9.
As outlined in the description of this experiment, we assembled a LVL2 plasmid possessing the Cas9 CDS as well as the gRNA with the dns spacer. In comparison with the cotransformation of the Cas9-Plasmid and the gRNA-plasmid, the transformation of one plasmid has a higher efficiency, especially when transformed in combination with the donor nucleooligotides.
For the knock-out of the dns in Vibrio natriegens, we cotransformed the LVL2 plasmid once with the suitable oligotides of the lagging strand, once with those of the leading strand and as a control once in the absence of the oligos. Figure 1 shows the plates of these transformations.
The control lacking the donor DNA showed no bacterial growth. This indicates that Cas9 is able to cleave the DNA in V. natriegens when coexpressed with gRNA, targeting the chromosome. This leads to the double strand break (DSB) of the DNA and cell death (Jiang et al.,2013).
The product of the transformation in which the donor nucleooligonucleotides were provided presented a strong bacterial growth on the plates. As was shown, the LVL2 plasmid is lethal to Vibrio. The addition of donor nucleooligotides facilitates the repair of the DSB through homologous recombination, thereby incorporating the desired mutation into the strand (Wright et al.,2013).

The picture shows colonies containing LVL2-Cas9-gRNA plasmid and A: no donor templates (no bacterial growth), B: donor templates of the leading strand and C: donor templates oft he lagging strand (both bacterial lawn)
This first experiment suggests that our CRIPSPR/Cas9 is able to generate mutations.
Our major goal was to accelerate the workflow of metabolic engineering. For that we tested enzymes from different organisms and could find the most promising candidates for our pathway. Additionaly we detected 3-hydroxypropionate via mass spectrometry by combining cell extracts of V. natriegens expressing Mcr or Acc. Beside that we constructed a rationally designed pathway and tested it in vitro. We were able to verify its functionality in V. natriegens by applying an enzyme activity assay. Furthermore, we successfully created a pathway library containing 390 pathway variants and cloned a biosensor for high throughput screening of 3HPA .
Enzyme activities of Acc from different organisms

The bar chart shows, that Acc + BirA from S. elongatus has the highest specific activity with 164 mU/mg, while Acc + BirA from E. coli has an activity of 14 mU/mg and wild type cells have no measurable activity.

The graph shows, that the absorption of NADPH at 365nm is constant. The initial mixture contained MOPS Buffer, MgCl2, ATP, NADPH and KHCO3. After addition of cell extract the absorption stays constant, but after addition of acetyl-CoA as start substrate, the absorption drops. The dashed line indicate the slopes that were used for calculating the specific activity. The two peaks are artifacts, induced by pipetting of reagents into the cuvette.
The specific activity was calculated via determination of absorption slopes (Fig 2). To ensure that the decrease of absorption is definitely due to malonyl-CoA addition and not addition of cell extract, we first added cell extract and saw no change of absorption. However, after malonyl-CoA addition the absorption instantly dropped. Therefore, we suppose that the measured NADPH consumption correlates with Acc activity. Unfortunately, we haven’t measured Acc activity from C. glutamicum yet because we built the corresponding plasmid library just recently and had no time for biochemical characterization. However, we measured a rationally designed construct bearing accBC, accD and birA from C. glutamicum and mcr from C. aurantiacus and were able to see activity (Fig. 6).
Mcr is soluble and functional in V. natriegens
The second enzyme in our pathway is the malonyl-CoA reductase (Mcr). The enzyme comes from C. aurantiacus, a thermophilic organism, therefore we wanted to test, if the Mcr is correctly expressed and functional in V. natriegens. For that we overexpressed it, lysed the cells via French press and made an SDS-PAGE as well as an enzyme activity assay.
On the SDS-PAGE we compared cell extract from wild type cells with extract from Mcr producing cells. Figure 3 shows, that the band for Mcr with a mass of 131 kDa is only visible in Mcr producing cells. This result confirms, that the protein is soluble and that Mcr production works.

In the enzyme activity assay, we also used cell extract from wild type cells and Mcr producing cells. Figure 4 shows a significant activity with extract of Mcr producing cells. The specific activity is 48 mU/mg. Therefore, we know now, that acetyl-CoA carboxylase, as well as malonyl-CoA reductase are both functional in our strain. The model predicted a higher activity of Mcr compared with Acc, but our measurements suggest the opposite. However, it isn’t possible to compare these data directly, because the calculations of specific activities depend on the protein amount and we used extract instead of purified proteins. If in the Acc extract the percentage of Acc is higher than the Mcr percentage in the Mcr extract, we got incomparable results.

So far, we could show, that Acc and Mcr are functional in V. natriegens and have sufficient activities. Next, we wanted to see if these enzymes can be combined to produce 3HPA . For that we combined cell extract from Mcr producing cells with extract from Acc producing cells and incubated the mixture with 400µM NADPH, 3.33mM ATP, 2mM Acetyl-CoA, 6.67mM MgCl2 and 50mM KHCO3 to let the reaction happen in vitro. To detect 3-hydroxypropionate we used triple quadrupole mass spectrometry (Fig. 5). To simultaneously test the hypothesis of our (metabolic model), that Acc is the rate limiting step in the route, we used two different ratios of Acc/Mcr. For each measurement we used the same amount of Mcr extract (6µl in a total volume of 50µl) and varied the amount of Acc extract (1.5µl/0.36µl). As a positive control we also used purified Acc (130µg/ml). We incubated the mixture for 30 minutes at 37°C, centrifuged it and used the supernatant for derivatization with 3-nitrophenyl-hydrazine (Han et al. 2013). To be able to quantify 3HPA later, we used samples with and without supplementation of a defined amount of 3HPA. For LC-MS we used in the first quadrupole a m/z filter of 224, then split the compound in the second quadrupole with a collision energy of 30V and for the third quadrupole we used a m/z filter of 194. All measurements were made with negative polarity. One important aspect is that we have to distinguish between 3HPA and the structural isomer lactate, which is also produced by V. natriegens (Hoffart et al. 2017). For the differentiation we added cell extract of V. natriegens wild type with 200µM 3HPA or 200µM lactate respectively. We came to the result that a good differentiation is possible by measuring the retention time, which was in our hands 2.947 minutes for 3HPA and 3.089 minutes for lactate.
 a
a b
b c
c d
dAll measurements were performed in a triple quadrupole mass spectrometer using the mass filters 224→194, a collision energy of 30V and negative polarity. We differentiated between 3HPA and lactate (a) and measured different amounts of cell extract, 1.5µl per 50µl reaction (b), 0.36µl (c) or purified propionyl-CoA carboxylase with D107I mutation, making the enzyme specific for acetyl-CoA carboxylation as a positive control (d).
Lactate has a longer retention time than 3HPA enabling a clear distinction. In the measurements of our cell extract samples, we could clearly identify 3HPA and see a peak at a retention time of 2.947 minutes. By comparing the ion counts of our extracts with the ion counts of our extracts with supplemented 3HPA we can calculate the amount of produced 3HPA, that can be seen in Table 1. We got 3HPA concentrations ranging from 92.48 to 453.34 µM. The fact that the first sample with 4-fold higher Acc extract amounts also has 4-fold higher 3HPA amounts shows the accuracy of the measurement but it also led us to the assumption, that the rate-limiting step in the reaction was the Acc abundance and that the 2mM Acetyl-CoA, that were injected at the beginning, weren’t consumed at all. If it would be consumed, the number of enzymes wouldn’t have played a role. Therefore, we recommend to repeat the assay and increase the incubation time at 37°C from 30 minutes to 2 hours or to photometrically determine the time point, when all the substrate is consumed. Nevertheless, we achieved to detect and quantify 3HPA in vitro with cell extracts of V. natriegens. Additionally, we developed a measuring protocol to differentiate between 3HPA and lactate. The next step will now be, to cotransform the plasmids for both enzymes into V. natriegens and determine in vivo production of 3HP.
| Sample | 3HPA concentration [µM] |
|---|---|
| 1.5µl Acc extract | 453.34 |
| 0.36µl Acc extract | 92.48 |
| Purified enzyme | 269.67 |
Construction and testing of our model-predicted pathway plasmid
One of our approaches was the construction of a rationally designed plasmid, bearing accBC, accD, birA and mcr with optimized expression levels, enabling an optimal flux. For that we implemented a model for a two-scale metabolic flux analysis, which can be seen in the Model section (link to model section). For that we first ordered the coding regions for these genes, codon optimized for V. natriegens and cloned them via Golden Gate into the level 0 backbone of the Marburg Collection. Then we used the toolbox to build level 1 plasmids, each of them bearing the transcriptional units, and finally combined them to a functional level 2 vector harbouring the whole pathway. Which parts we used can be seen in the Rational Design section of Metabolic Engineering (link to design section). To investigate pathway performance, we transformed the plasmid into V. natriegens, lysed the cells and used them for an activity assay, starting with Acetyl-CoA (Figure 6). The data show that cells that contain the pathway plasmid consume NADPH a lot faster, indicating that the pathway is working and 3-hydroxypropionate is produced. The specific activity is 0.025 mU/mg, what is low in comparison to the activities of our single enzyme measurements. We think a major reason for that is the quality of the cell extract. After preparing it we stored it at -20° for about two weeks because we had many other experiments simultaneously we primarily had to focus on. When cell extract is not fresh, its activity can drop rapidly. Nevertheless, the assay showed that there is activity, so our pathway works in V. natriegens. The next step to get better results would be to repeat the assay with fresh extract. It would also be useful to let the reaction happen and then use LC-MS or our malonyl-CoA sensor to screen for malonyl-CoA. If there is detectable malonyl-CoA, then the pathway flow is not optimal, because malonyl-CoA should not accumulate.
Construction of the pathway library

The bar plot shows the specific activities of cell extract from V. natriegens with and without our rationally designed pathway plasmid. The result is that wild type cells have an activity of 0.003 mU/mg, while extract of cells performing the pathway has a specific activity of 0.025, showing that the pathway works and produces 3-hydroxypropionate.
So far, we showed, that the Acc, as well as the Mcr are both soluble and functional in V. natriegens and we were able to prove 3HPA production in cell extracts. Furthermore, we built a rationally designed pathway and implemented it into V. natriegens. However, our aim was not only to establish one pathway but rather a whole library of pathway variations. As mentioned in the Pathway Library section of Metabolic Engineering (link to design section), we planned to construct 1446 pathway variants. Since the cloning of so many constructs is nearly unrealistic, we used the liquid transfer robot Echo 525 Liquid Handler from Labcyte to make the cloning. Although the robot works very fast and is able to mix all golden gate reactions in less than one hour, we wanted to start with a shorter number of reactions, to test if our strategy works. Nobody has ever tested golden gate cloning coupled with natural transformation in that robot. Therefore, we focused on one 96 well plate for each plasmid category. We actually planned and performed the cloning of all 24 constructs of category I, 96 constructs of category II, 96 constructs of category III, all 81 constructs of category IV, all 27 constructs of category V, all 81 constructs of category VI and all 81 constructs of category VII, yielding 486 constructs in total. For the cloning we prepared a 384 well source plate containing each part of the Marburg Collection. We then prepared a picking list for the robot and it started to transfer all 486 golden gate mixtures into 96 well plates. A more detailed description of the Echo Liquid Handler is specified in the Labautomation-section [Link zu labautomation]. The overview about all plasmids is shown in the Pathway library section of Metabolic Engineering (link to design section). After running the golden gate reaction in a thermocycler, we used natural transformation to transform the constructs into V. natriegens. For that we added Tfox-bearing cells to each well, performed the protocol for natural transformation, that is shown in the Methods section of Strain Engineering ( link to design section) and plated them out. To our pleasure 390 plates showed colonies after transformation. Theoretically, the next step would be to make minipreps and check plasmid correctness, but according to our metabolic engineering workflow, we would prefer to directly use the biosensor to test how efficient the pathway variants are. At the end the lack of time hindered us from testing the pathway library, but our previous cloning approaches with the Marburg Collection had efficiencies of at least 80%, therefore we assume that at least 312 constructs out of these 390 are correct, which is still a high number and sufficient to continue with biosensor-guided product screening and directed evolution to optimize selected pathway variants in the future.
Biosensors
In the first major experiment we tested induction of lux expression over a 3-HPA gradient. When we first saw the plate reader data, we were very optimistic that we had successfully cloned a functional 3-HPA sensor into V. natriegens. Just from the luminescence values, a clear trend to more emission at medium concentrations was apparent.
To explain this, we argued that a higher concentration of 3-HPA caused so much stress, the cells died, and that lower concentrations were insufficient for induction. The difference between the uninduced and the most highly expressing sample was about six-fold.

B) The concentration of 3HPA plotted versus the luminescence
What we also observed was a severe loss of vitality, materializing in form of significantly reduced growth which completely ceased at the two highest concentrations, 10 mM and 6.6 mM respectively. This would represent a significantly lower resilience to 3-HPA than previously reported for E. coli (Liu et al. 2016). There, the threshold at which first detrimental effects could be seen was 200 mM. If we want to make industrial production of 3-HPA in V. natriegens a reality, a way to increase its resistance is indispensable. Fortunately, strategies to implement such increased resilience have already been shown to work in E. coli (Liu et al. 2016). So, given the time, similar solutions for V. natriegens are not hard to imagine.
Several different factors could be responsible for the toxicity. At first, we thought that 3-HPA's structural similarity to cellular substrates, like for instance lactate, could cause inhibition of enzymes acting on these substrates. The mechanism of toxicity for 3-HPA is most likely its acidity. 3-HPA is a weak acid with a pKs of 4.50, and in our basic research (link to protocol for pH tolerance assay) we showed that a low pH does indeed slow the growth of V. natriegens, or can even be lethal. This could be compensated in part by choosing an alkaline starting pH of the culture media. A step that is favored, as V. natriegens is less susceptible to a high pH up to 9.5.
However, this does not explain the higher luminescence for medium concentrations, where growth does not appear to be impacted.
When we took a closer look at our control, the lux operon driven by J23107 ( http://parts.igem.org/Part:BBa_J23107 ), we found the same pattern of dead cells at high concentrations, high lux expression in the middle and background expression at lower concentrations. J23107 is a constitutive promoter of medium strength, chosen to be close to the expression levels we saw from our uninduced sensor. Regrettably, this shows that our biosensor is not functional, but presents us with an interesting enigma to solve.
It is plausible that 3-HPA , which is very similar to some substrates for enzymes of the lux operon, could in some way interact with said enzymes. Another explanation voiced by a team member is based on the fact that through division, cells dilute their cell content. This includes metabolites, DNA and proteins. If the cells divide more slowly, they have more time to accumulate these. To accumulate the proteins responsible for lux luminescence, which would result in a higher per OD luminescence. Our observations do not quite fit this, since we not only see a higher luminescence per OD, but a much higher luminescence overall. Also conceivable is, that resources that would normally go into cell growth are diverted into lux expression.

There are no sources to substantiate that claim, but we found another promising lead in a source on the behavior of lux expression depending on the pH (Dorn et al. 2003). There, it was shown that pH is a major influence in lux expression. They observed the peak luminescence at a slightly acidic pH of around 6. This is remarkably close to the values we measured for the concentrations of 1.9 mM to 2.9 mM, where we also observed the strongest luminescence. Therefore, when luminescence is influenced that strongly by pH, that explains why our constitutively expressed lux reporter showed the same pattern. Amazingly, this effect was already apparent only five minutes after induction.
For the malonyl-CoA sensor, we had difficulties to clone the level 2 plasmids required for them to work. Once we solved this issue, testing may proceed.
There are several other sensors for 3-HPA as well as for malonyl-CoA as discussed in our design section.
One promising way to directly screen for productivity of our 3-HPA pathway would be to co-express the lux operon under a strong constitutive promoter. Since the substrates for the luminescence reaction are part of the fatty acid synthesis pathway, and malonyl-CoA is at the root of this pathway, a higher conversion rate of malonyl-CoA to 3-HPA should diminish luminescence output. This simple competitive assay could give a rudimentary indication of the Mcr activity.
Directed evolution
After building a pathway library, transferring it into V. natriegens and establishing biosensors, the next step in our metabolic engineering workflow is to implement directed evolution for pathway optimization. So far, we could show that our enzymes are functional and 3HPA is produced, but there are still many possibilities to optimize this metabolic pathway. Unfortunately, lack of time forced us to skip the introduction of evolution into our approach. Nevertheless, we planned the whole procedure so that everyone can start directly with the implementation. As already mentioned in the Design section of metabolic engineering
, we followed two strategies. The first one is to re-feed 3HPA into the central metabolism and forcing the strain to perform this metabolic route. For that we decided to introduce genes for a propionyl-CoA synthase (Pcs), a propionyl-CoA carboxylase (Pcc), a methylmalonyl-CoA epimerase and a methylmalonyl-CoA mutase. Pcs converts 3HPA into propionyl-CoA, which then gets carboxylated by Pcc to form (S)-2-methylmalonyl-CoA. Methylmalonyl-CoA epimerase and methylmalonyl-CoA mutase finally catalyze the isomerization of (S)-2-methylmalonyl-CoA via (R)-2-methylmalonyl-CoA to succinyl-CoA. By deleting housekeeping genes for succinyl-CoA production, the only possibility for the cell to produce this compound is by using our pathway. Coupled with the fast growth of V. natriegens, cells will fast adapt to this route and naturally optimize it. To start with this strategy, the next step would be to design oligos for genomic deletion of succinyl-CoA production. Then MuGent can be used to delete these genes, as shown in the Methods section of Strain Engineering ( link to design section). Afterwards, the genes for our bypass can be introduced and directed evolution is started. For our second strategy, where we use FACS and MAGE to introduce genome modifications, we already looked for possible modifications, we could introduce via oligos. A table of that modifications is shown in the Further Improved section of Metabolic Engineering ( link to design section). Here the next steps would be to compare the list of modified genes from other organisms with V. natriegens and design oligos for these modifications. After calibrating the biosensor one can start with MAGE-directed optimization coupled with FACS-directed selection of promising cells.
References
Dorn, J.G., Frye, R.J., Maier, R.M., 2003. Effect of temperature, pH, and initial cell number on luxCDABE and nah gene expression during naphthalene and salicylate catabolism in the bioreporter organism Pseudomonas putida RB1353. Appl. Environ. Microbiol. 69, 2209-16Han, J., Gagnon, S., Eckle, T., Borchers, C.H., 2013. Metabolomic analysis of key central carbon metabolism carboxylic acids as their 3-nitrophenylhydrazones by UPLC/ESI-MS. Electrophoresis 34, 2891–2900. https://doi.org/10.1002/elps.201200601
Hanko, E.K.R., Minton, N.P., Malys, N., 2017. Characterisation of a 3-hydroxypropionic acid-inducible system from Pseudomonas putida for orthogonal gene expression control in Escherichia coli and Cupriavidus necator
Hoffart, E., Grenz, S., Lange, J., Nitschel, R., Müller, F., Schwentner, A., Feith, A., Lenfers-Lücker, M., Takors, R., Blombach, B., 2017. High substrate uptake rates empower Vibrio natriegens as production host for industrial biotechnology. Appl. Environ. Microbiol. 83. https://doi.org/10.1128/AEM.01614-17
Liu, M., Han, X., Xian, M., Ding, Y., Liu, H., Zhao, G., 2016. Development of a 3-hydroxypropionate resistant Escherichia coli strain. Blokesch, M., Schoolnik, G. K., The Extracellular Nuclease Dns and Its Role in Natural Transformation of Vibrio cholerae, JOURNAL OF BACTERIOLOGY, (2008), 7232–7240.
D'Ari, R., A. Jaffe, P. Bouloc & A. Robin, (1988) Cyclic AMP and cell division in Escherichia coli. Journal of bacteriology 170: 65-70. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC210606/
Dalia TN, Hayes CA, Stolyar S, Marx CJ, McKinlay JB, Dalia AB. Multiplex Genome Editing by Natural Transformation (MuGENT) for Synthetic Biology in Vibrio natriegens. ACS Synth Biol. (2017) 15;6(9):1650-1655.
Hoffart E, Grenz S, Lange J, Nitschel R, Müller F, Schwentner A, Feith A, Lenfers-Lücker M, Takors R, Blombach B2. (2017) High substrate uptake rates empower Vibrio natriegens as production host for industrial biotechnology. Appl Environ Microbiol. (2017) 01614-17. https://www.ncbi.nlm.nih.gov/pubmed/28887417
Weinstock, M. T., E. D. Hesek, C. M. Wilson and D. G. Gibson (2016). "Vibrio natriegens as a fast-growing host for molecular biology." Nat Methods 13(10): 849-851.
Zeaiter, Z., Mapelli F., Crotti, E., Borin, S. Methods for the genetic manipulation of marine bacteria. Electronic Journal of Biotechnology. (2018) 33: 17-28.