|
|
| Line 48: |
Line 48: |
| | <br> | | <br> |
| | </p> | | </p> |
| − | <img style="float:right; width:400px;margin: 1% "src="https://static.igem.org/mediawiki/2018/7/79/T--Marburg--cycle_cooper.png">
| + | This wordplay by Sir Arthur Eddington highlights the importance of math-based models in every discipline of science. In Physics, the incredible potential of math has long been known and utilized. Chemistry was lacking behind a bit, but there is an incredible theoretical branch of chemistry that took a lot out of methods rooted in physics. Biology is, sadly, lacking behind its sister sciences. |
| − | This wordplay by Sir Arthur Eddington highlights the importance of math-based models in every discipline of science. In Physics, the incredible potential of math has long been known and utilized. Chemistry was lacking behind a bit, but there is an incredible theoretical branch of chemistry that took a lot out of methods rooted in physics. Biology is, sadly, lacking behind its sister sciences.
| + | Every theory and every model only realizes its full potential with experimental confirmation while experimental results can be achieved without any model. But in conjunction with each other, the boundaries of science can be heightened dramatically. |
| − | Every theory and every model only realizes its full potential with experimental confirmation while experimental results can be achieved without any model. But in conjunction with each other, the boundaries of science can be heightened dramatically.
| + | <br> |
| − | <br>
| + | Our Project is centered around saving time, streamlining processes and offering these advantages to the community. |
| − | Our Project is centered around saving time, streamlining processes and offering these advantages to the community.
| + | We improved processes to run faster, but another way that should be implemented in parallel is predicting what to test in the lab and therefore eliminating unnecessary experiments. |
| − | We improved processes to run faster, but another way that should be implemented in parallel is predicting what to test in the lab and therefore eliminating unnecessary experiments.
| + | The most powerful process that allows for improved prediction power or is the reason that prediction is even possible is modeling. |
| − | The most powerful process that allows for improved prediction power or is the reason that prediction is even possible is modeling.
| + | We harvested this prediction power with two independent modeling approaches, one predicting the metabolism of Vibrio Natriegens and the other designing an enzyme capable of a novel reaction, decarboxylating malate to 3-hydroxypropionic acid ( <dfn data-info="3-hydroxypropionic acid">3HPA</dfn>). |
| − | We harvested this prediction power with two independent modeling approaches, one predicting the metabolism of Vibrio Natriegens and the other designing an enzyme capable of a novel reaction, decarboxylating malate to 3-hydroxypropionic acid ( <dfn data-info="3-hydroxypropionic acid">3HPA</dfn>).
| + | |
| | | | |
| | | | |
| Line 62: |
Line 61: |
| | | | |
| | <div style="display: inline-block; align-items: center;"> | | <div style="display: inline-block; align-items: center;"> |
| − | <div style="float:left; width: 49%; text-align: justify;">
| + | <div style="float:left; width: 49%; text-align: justify;"> |
| − | <h2>Metabolic Model</h2>
| + | <h2>Metabolic Model</h2> |
| − | <br>
| + | <br> |
| − | In order to be able to fine-tune synthetic pathways and utilize the well-characterized parts of our cloning toolbox to its fullest potential, we need a model which gives useful predictions of metabolic fluxes. There exists a multitude of approaches to model fluxes – some are on the genomic scale some occupied only with the mechanism of a single enzyme.
| + | In order to be able to fine-tune synthetic pathways and utilize the well-characterized parts of our cloning toolbox to its fullest potential, we need a model which gives useful predictions of metabolic fluxes. There exists a multitude of approaches to model fluxes – some are on the genomic scale some occupied only with the mechanism of a single enzyme. |
| − | We have chosen to utilize two different approaches – on one scale we use reaction kinetics to analyze the behavior/kinetics of our inserted enzymes within the metabolism – on the other we use a novel approach called 2S-MFA [Reference] to get a genome-scale perspective on the metabolic fluxes.
| + | We have chosen to utilize two different approaches – on one scale we use reaction kinetics to analyze the behavior/kinetics of our inserted enzymes within the metabolism – on the other we use a novel approach called 2S-MFA [Reference] to get a genome-scale perspective on the metabolic fluxes. |
| − | Using the thereby consolidated theoretical knowledge we can complete the Design-Build-Test-Learn cycle for our metabolic engineering project and use it to iteratively improve our project. With the prediction power of this models, we not only save a lot of time in the lab but we manage to improve the efficiency and productivity, the skill ceiling of our pathway.
| + | Using the thereby consolidated theoretical knowledge we can complete the Design-Build-Test-Learn cycle for our metabolic engineering project and use it to iteratively improve our project. With the prediction power of this models, we not only save a lot of time in the lab but we manage to improve the efficiency and productivity, the skill ceiling of our pathway. |
| | | | |
| − | </div>
| + | </div> |
| − | <div style="float:right; width: 49%; text-align: justify;">
| + | <div style="float:right; width: 49%; text-align: justify;"> |
| − | <h2>Structural Model</h2>
| + | <h2>Structural Model</h2> |
| − | <br>
| + | <br> |
| − | The pathway we used for production of <dfn data-info="3-hydroxypropionic acid">3HPA</dfn> (LINK ZU Metabolic) has been explored previously and is based on a combination of known reactions and known enzymes.
| + | The pathway we used for production of <dfn data-info="3-hydroxypropionic acid">3HPA</dfn> (LINK ZU Metabolic) has been explored previously and is based on a combination of known reactions and known enzymes. |
| − | Combination of different enzymes to make a synthetic pathway is a well-established method in the field of metabolic engineering, but limited to existing reactions and known enzymes.
| + | Combination of different enzymes to make a synthetic pathway is a well-established method in the field of metabolic engineering, but limited to existing reactions and known enzymes. |
| − | With our structural model, we tried to build a new energetically more favorable pathway which was previously impossible.
| + | With our structural model, we tried to build a new energetically more favorable pathway which was previously impossible. |
| − | To accomplish that, we needed to implement a reaction with no known enzyme to catalyze it, the decarboxylation reaction of malate to (<dfn data-info="3-hydroxypropionic acid">3HPA</dfn>).
| + | To accomplish that, we needed to implement a reaction with no known enzyme to catalyze it, the decarboxylation reaction of malate to (<dfn data-info="3-hydroxypropionic acid">3HPA</dfn>). |
| − | To build this pathway we decided to engineer an enzyme capable of catalyzing this reaction.
| + | To build this pathway we decided to engineer an enzyme capable of catalyzing this reaction. |
| − | We performed a literature research and developed an idea on how to build a binding pocket catalyzing this reaction.
| + | We performed a literature research and developed an idea on how to build a binding pocket catalyzing this reaction. |
| − | To evaluate if our binding pocket works, we performed electronic structure calculations.
| + | To evaluate if our binding pocket works, we performed electronic structure calculations. |
| − | With that, we were able to calculate the activation barrier of the reaction and therefore evaluate if it is possible for the reaction to take place.
| + | With that, we were able to calculate the activation barrier of the reaction and therefore evaluate if it is possible for the reaction to take place. |
| − | To advance from a binding pocket to a full enzyme we evaluated <i>in silico</i> mutated versions of acetolactate decarboxylase (<dfn data-info="Acetolactate Decarboxylase">ALD</dfn>).
| + | To advance from a binding pocket to a full enzyme we evaluated <i>in silico</i> mutated versions of acetolactate decarboxylase (<dfn data-info="Acetolactate Decarboxylase">ALD</dfn>). |
| − | For evaluating which mutants perform best we used <dfn data-info="Molecular Dynamics">MD</dfn> simulations and checked how well the binding pocket assumed in the electronic structure calculations is represented in the simulations.
| + | For evaluating which mutants perform best we used <dfn data-info="Molecular Dynamics">MD</dfn> simulations and checked how well the binding pocket assumed in the electronic structure calculations is represented in the simulations. |
| − | With the help of these <i>in silico</i> approaches we chose a mutant and tested it in the wetlab.
| + | With the help of these <i>in silico</i> approaches we chose a mutant and tested it in the wetlab. |
| − | </div>
| + | </div> |
| − | </div>
| + | </div> |
| | <div class="collapsible"> | | <div class="collapsible"> |
| − | <div class="btn_expand">Metabolic Model</div>
| + | <div class="btn_expand">Metabolic Model</div> |
| − | <div class="content">
| + | <div class="content"> |
| − | Some Stuff from Andrej
| + | Some Stuff from Andrej |
| − | </div>
| + | </div> |
| | </div> | | </div> |
| | | | |
| | | | |
| | <div class="collapsible"> | | <div class="collapsible"> |
| − | <div class="btn_expand">Structural Model</div>
| + | <div class="btn_expand">Structural Model</div> |
| − | <div class="content">
| + | <div class="content"> |
| − | <h2>Teach the rabbit to quack <br> Expanding the scope of Metabolic Engineering through novel enzyme engineering</h2>
| + | <h2>Teach the rabbit to quack <br> Expanding the scope of Metabolic Engineering through novel enzyme engineering</h2> |
| | <h3>Infrastructure</h3> | | <h3>Infrastructure</h3> |
| | <p> | | <p> |
| − | We want to thank Prof. Klebe and Prof. Kolb for kindly providing us with the necessary infrastructure and software access to perform our calculations.
| + | We want to thank Prof. Klebe and Prof. Kolb for kindly providing us with the necessary infrastructure and software access to perform our calculations. |
| − | For our <dfn data-info="Quantum Mechanics">QM</dfn> calculations, we used the program GAUSSIAN09 and Chemcraft to set up our systems.
| + | For our <dfn data-info="Quantum Mechanics">QM</dfn> calculations, we used the program GAUSSIAN09 and Chemcraft to set up our systems. |
| − | For a foray to how these kinds of calculations work, please click here.
| + | For a foray to how these kinds of calculations work, please click below. |
| | | | |
| − | <div class="collapsible">
| + | <div class="collapsible"> |
| − | <div class="btn_expand">Foray to <dfn data-info="Quantum Mechanics">QM</dfn> Calculations</div>
| + | <div class="btn_expand">Foray to <dfn data-info="Quantum Mechanics">QM</dfn> Calculations</div> |
| − | <div class="content">
| + | <div class="content"> |
| − | <h3>Foray to <dfn data-info="Quantum Mechanics">QM</dfn> Calculations [<a href=""><abbr title=""></a>]</h3>
| + | <h3>Foray to <dfn data-info="Quantum Mechanics">QM</dfn> Calculations [<a href=""><abbr title=""></a>]</h3> |
| − | <p>
| + | <p> |
| − | <dfn data-info="Quantum Mechanics">QM</dfn> Calculations are the pinnacle of precision compared to all other methods used for calculating molecular systems, but this precision comes with very high computational costs.
| + | <dfn data-info="Quantum Mechanics">QM</dfn> Calculations are the pinnacle of precision compared to all other methods used for calculating molecular systems, but this precision comes with very high computational costs. |
| − | What the computer actually does when doing this type of calculations is calculating the Hamiltonian of the system and solving the Schroedinger Equation that way.
| + | What the computer actually does when doing this type of calculations is calculating the Hamiltonian of the system and solving the Schroedinger Equation that way. |
| − | With the help of this, we can describe the system.
| + | With the help of this, we can describe the system. |
| − | <dfn data-info="Quantum Mechanics">QM</dfn> Calculations are characterized by two important things, the functional and the basis set used.
| + | <dfn data-info="Quantum Mechanics">QM</dfn> Calculations are characterized by two important things, the functional and the basis set used. |
| − | The functional is characterized by the way the Hamiltonian is calculated.
| + | The functional is characterized by the way the Hamiltonian is calculated. |
| − | For all calculations that we do, we also need a starting point to describe orbitals or the density function (We will go into more detail to this later) and the basis set describes the number and type of the orbital functions that we use to calculate the system.
| + | For all calculations that we do, we also need a starting point to describe orbitals or the density function (We will go into more detail to this later) and the basis set describes the number and type of the orbital functions that we use to calculate the system. |
| − | </p>
| + | </p> |
| − | <h3>Born Oppenheimer Approximation</h3>
| + | <h3>Born Oppenheimer Approximation</h3> |
| − | <p>
| + | <p> |
| − | A necessary assumption to use <dfn data-info="Quantum Mechanics">QM</dfn> as well as <dfn data-info="Molecular Mechanics">MM</dfn>
| + | A necessary assumption to use <dfn data-info="Quantum Mechanics">QM</dfn> as well as <dfn data-info="Molecular Mechanics">MM</dfn> |
| | methods is the Born Oppenheimer Approximation. | | methods is the Born Oppenheimer Approximation. |
| − | This is the approximation that due to the vast difference in speed between the electrons and the nucleus of the atoms the movement of both can be investigated apart from each other.
| + | This is the approximation that due to the vast difference in speed between the electrons and the nucleus of the atoms the movement of both can be investigated apart from each other. |
| − | For <dfn data-info="Quantum Mechanics">QM</dfn> Methods this means that we only look at the movement of electrons and neglect the very slow movements of the nuclei when solving the Schroedinger Equation.
| + | For <dfn data-info="Quantum Mechanics">QM</dfn> Methods this means that we only look at the movement of electrons and neglect the very slow movements of the nuclei when solving the Schroedinger Equation. |
| − | </p>
| + | </p> |
| − | <h3>Functionals and Basis sets</h3>
| + | <h3>Functionals and Basis sets</h3> |
| − | <h3>Functionals</h3>
| + | <h3>Functionals</h3> |
| − | <p>
| + | <p> |
| − | There are two fundamentally different type of functionals, wave function based and density function based methods.
| + | There are two fundamentally different types of functionals, wave function based and density function based methods. |
| − | We only used density function based methods, to be precise B3Lyp.
| + | We only used density function based methods, to be precise B3Lyp. |
| − | Density Function Theory (DFT) is centered not on the Wavefunction but on the square of the wave function, the electron density.
| + | Density Function Theory (DFT) is centered not on the Wavefunction but on the square of the wave function, the electron density. |
| − | It is based on the theorem of Hohenberg and Kohn and modern DFT is also based on the Kohn Sham approach.
| + | It is based on the theorem of Hohenberg and Kohn and modern DFT is also based on the Kohn Sham approach. |
| − | The electron density is a measurable quantity that is dependent on the three vectors of spaces.
| + | The electron density is a measurable quantity that is dependent on the three vectors of spaces. |
| − | In the first days of DFT (when invented by physicists) mostly the Linear Density Approach (LDA) was used and this approach works fine for metals, in which the electrons are evenly distributed inside the metal.
| + | In the first days of DFT (when invented by physicists) mostly the Linear Density Approach (LDA) was used and this approach works fine for metals, in which the electrons are evenly distributed inside the metal. |
| − | However, this is a poor approximation once we want to model molecular systems.
| + | However, this is a poor approximation once we want to model molecular systems. |
| − | As a further Development of this approximation, Gradient corrected methods (GGA) have been invented.
| + | As a further Development of this approximation, Gradient corrected methods (GGA) have been invented. |
| − | These methods consider the electron density not to be uniform.
| + | These methods consider the electron density not to be uniform. |
| − | <br>
| + | <br> |
| − | There are multiple terms in the Hamiltonian that have to be calculated.
| + | There are multiple terms in the Hamiltonian that have to be calculated. |
| − | The most troublesome one is the so-called "Exchange-Correlation" term.
| + | The most troublesome one is the so-called "Exchange-Correlation" term. |
| − | There is a class of DFT functionals, the so-called Hybrid Functionals, that use a combination of exchange-correlation terms of different methods.
| + | There is a class of DFT functionals, the so-called Hybrid Functionals, that use a combination of exchange-correlation terms of different methods. |
| − | The method we used, B3LYP, uses a combination of LDA, GGA and Hartree Fock (A wavefunction based method) exchange-correlation.
| + | The method we used, B3LYP, uses a combination of LDA, GGA, and Hartree Fock (A wavefunction based method) exchange-correlation. |
| | | | |
| − | </p>
| + | </p> |
| − | <h3>Basis Sets</h3>
| + | <h3>Basis Sets</h3> |
| − | <p>
| + | <p> |
| − | Now that we know which method we use we can take a look at the second important characteristic of our <dfn data-info="Quantum Mechanics">QM</dfn> calculations, the basis set.
| + | Now that we know which method we use we can take a look at the second important characteristic of our <dfn data-info="Quantum Mechanics">QM</dfn> calculations, the basis set. |
| − | All methods described previously, even if based on DFT, use Orbitals in their calculations.
| + | All methods described previously, even if based on DFT, use Orbitals in their calculations. |
| − | By combining a certain number of Orbitals the orbitals of the system have to be expressed.
| + | By combining a certain number of Orbitals the orbitals of the system have to be expressed. |
| − | This starting number of orbitals is called the basis set.
| + | This starting number of orbitals is called the basis set. |
| − | Obviously, the more orbitals we have the more accurate the calculations become, but they also get more demanding.
| + | Obviously, the more orbitals we have the more accurate the calculations become, but they also get more demanding. |
| − | As a small basis set we used the 6-31g variant and as a bigger basis set, we used cc-pVDZ.
| + | As a small basis set we used the 6-31g variant and as a bigger basis set, we used cc-pVDZ. |
| − | </p>
| + | </p> |
| − | <h3>Summary</h3>
| + | <h3>Summary</h3> |
| − | <ul>
| + | <ul> |
| − | <li>Based on Quantum Mechanics</li>
| + | <li>Based on Quantum Mechanics</li> |
| − | <li>All electrons investigated</li>
| + | <li>All electrons investigated</li> |
| − | <li>Bond cleavage and formation can be calculated</li>
| + | <li>Bond cleavage and formation can be calculated</li> |
| − | <li>Slow Calculations and limited to small Systems</li>
| + | <li>Slow Calculations and limited to small Systems</li> |
| − | </ul>
| + | </ul> |
| − | </div>
| + | </div> |
| | </div> | | </div> |
| − | To perform <dfn data-info="Molecular Dynamics">MD</dfn> Simulations we used the program package AMBER.
| + | To perform <dfn data-info="Molecular Dynamics">MD</dfn> Simulations we used the program package AMBER. |
| − | For a foray how <dfn data-info="Molecular Dynamics">MD</dfn> Simulations work, please click here.
| + | For a foray how <dfn data-info="Molecular Dynamics">MD</dfn> Simulations work, please click below. |
| | <div class="collapsible"> | | <div class="collapsible"> |
| − | <div class="btn_expand">Foray to <dfn data-info="Molecular Dynamics">MD</dfn> Simulations</div>
| + | <div class="btn_expand">Foray to <dfn data-info="Molecular Dynamics">MD</dfn> Simulations</div> |
| − | <div class="content">
| + | <div class="content"> |
| − | <h3>Foray to Molecular Mechanics </h3>
| + | <h3>Foray to Molecular Mechanics </h3> |
| − | <p>
| + | <p> |
| | Molecular Mechanics (<dfn data-info="Molecular Mechanics">MM</dfn> ) uses the laws of classical mechanics to model molecular systems. It can be used to calculate anything ranging between small molecules to big proteins. | | Molecular Mechanics (<dfn data-info="Molecular Mechanics">MM</dfn> ) uses the laws of classical mechanics to model molecular systems. It can be used to calculate anything ranging between small molecules to big proteins. |
| | Each atom is simulated as one single particle, with a radius and charge. | | Each atom is simulated as one single particle, with a radius and charge. |
| − | Bonded interactions are treated by the famous Hookes law (i.e. they are springs). | + | Bonded interactions are treated by the famous Hooke's law (i.e. they are springs). |
| | This approximation allows us to simulate very big systems, but it is very important to asses which properties we can simulate correctly and therefore investigate. | | This approximation allows us to simulate very big systems, but it is very important to asses which properties we can simulate correctly and therefore investigate. |
| | Because classical Molecular Dynamics (<dfn data-info="Molecular Dynamics">MD</dfn>) Simulations are based on <dfn data-info="Molecular Mechanics">MM</dfn> | | Because classical Molecular Dynamics (<dfn data-info="Molecular Dynamics">MD</dfn>) Simulations are based on <dfn data-info="Molecular Mechanics">MM</dfn> |
| Line 234: |
Line 233: |
| | <p> | | <p> |
| | <dfn data-info="Molecular Dynamics">MD</dfn> Simulations are performed within a confined volume, called a unit cell. | | <dfn data-info="Molecular Dynamics">MD</dfn> Simulations are performed within a confined volume, called a unit cell. |
| − | This unit cell contains all particles that we simulate (i.e. protein,water,ligand) | + | This unit cell contains all particles that we simulate (i.e. protein, water, ligand) |
| | This introduces the problem of boundary effects because atoms and molecules close to these boundaries of the unit cell have fewer interaction partners than those in the middle of it. | | This introduces the problem of boundary effects because atoms and molecules close to these boundaries of the unit cell have fewer interaction partners than those in the middle of it. |
| | To avoid boundary effects at the edges of the unit cell we repeat the unit cell periodically. | | To avoid boundary effects at the edges of the unit cell we repeat the unit cell periodically. |
| | Thus, the shape of the unit cell has to allow such that a regular space filling lattice of unit cells can be arranged. | | Thus, the shape of the unit cell has to allow such that a regular space filling lattice of unit cells can be arranged. |
| − | In our study we used a truncated octahedron. | + | In our study, we used a truncated octahedron. |
| | <figure> | | <figure> |
| | <img style="float:right; height:400px;" src="https://upload.wikimedia.org/wikipedia/commons/7/7c/Truncatedoctahedron.gif" alt="text"> | | <img style="float:right; height:400px;" src="https://upload.wikimedia.org/wikipedia/commons/7/7c/Truncatedoctahedron.gif" alt="text"> |
| Line 253: |
Line 252: |
| | <li>Fast Calculations and big Systems</li> | | <li>Fast Calculations and big Systems</li> |
| | </ul> | | </ul> |
| − | </div>
| + | </div> |
| | </div> | | </div> |
| − | <div class="skipTarget" skipname="Motivation Structural Model"></div>
| + | <div class="skipTarget" skipname="Motivation Structural Model"></div> |
| | <h3>Motivation</h3> | | <h3>Motivation</h3> |
| | <p> | | <p> |
| − | We went to great lengths to develop a workflow for metabolic engineering that utilizes the incredible power of directed evolution and is worthy of synthetic biology in the not so early 21st Century.
| + | We went to great lengths to develop a workflow for metabolic engineering that utilizes the incredible power of directed evolution and is worthy of synthetic biology in the not so early 21st Century. |
| − | With the very recent Nobel prize in chemistry towards directed evolution we think we have struck a nerve, and with <i>Vibrio Natriegens</i> this process can be streamlined even further.
| + | With the very recent Nobel prize in chemistry towards directed evolution we think we have struck a nerve, and with <i>Vibrio Natriegens</i> this process can be streamlined even further. |
| − | Traditional Chemistry with Synthesis as its supreme discipline and its implications in industry is one of the driving forces behind the modern wealth and is being improved on a daily basis.
| + | Traditional Chemistry with Synthesis as its supreme discipline and its implications in the industry is one of the driving forces behind the modern wealth and is being improved on a daily basis. |
| − | Synthetic Biology and in particular Metabolic Engineering as a chemical producing science can use that incredible knowledge and expand on it.
| + | Synthetic Biology and in particular Metabolic Engineering as a chemical producing science can use that incredible knowledge and expand on it. |
| − | Due to the completely different way of synthesis, we can mend the problems that classical synthesis is facing (e.g. natural products with many stereocenters, neccessary purification of products after most reaction steps) and synthesize compounds previously not able to be synthesized (e.g. Artemisinic acid) or ones that were too costly.
| + | Due to the completely different way of synthesis, we can mend the problems that classical synthesis is facing (e.g. natural products with many stereocenters, necessary purification of products after most reaction steps) and synthesize compounds previously not able to be synthesized (e.g. Artemisinic acid) or ones that were too costly. |
| − | Using metabolic engineering also has the advantage to produce chemical out of renewable resources while many chemicals right now are synthesized starting with fossil oil.
| + | Using metabolic engineering also has the advantage to produce chemical out of renewable resources while many chemicals right now are synthesized starting with fossil oil. |
| − | To further improve this incredible opportunity to make more of the chemical space synthesizable in a cheap, easy and renewable manner we need to go beyond "mix and match" pathways and explore novel enzymatic reactions.
| + | To further improve this incredible opportunity to make more of the chemical space synthesizable in a cheap, easy and renewable manner we need to go beyond "mix and match" pathways and explore novel enzymatic reactions. |
| − | According to
| + | According to |
| | | | |
| − | <a href="https://www.sciencedirect.com/science/article/pii/S1367593116302071"><abbr title ="Tobias J Erb, Patrik R Jones, Arren Bar-Even, Synthetic metabolism: metabolic engineering meets enzyme design, Current Opinion in Chemical Biology (2017) 56-62" >(Erb <i>et al.</i>2017)</abbr></a>
| + | <a href="https://www.sciencedirect.com/science/article/pii/S1367593116302071"><abbr title ="Tobias J Erb, Patrik R Jones, Arren Bar-Even, Synthetic metabolism: metabolic engineering meets enzyme design, Current Opinion in Chemical Biology (2017) 56-62" >(Erb <i>et al.</i>2017)</abbr></a> |
| | | | |
| − | metabolic engineering has been categorized in 5 different levels, depending on the methods employed.
| + | metabolic engineering has been categorized in 5 different levels, depending on the methods employed. |
| − | These levels and the corresponding metabolic space is displayed in Figure [1].
| + | These levels and the corresponding metabolic space is displayed in Figure 1. |
| − | We tried to expand on what we already did in our metabolic engineering project and tap the huge advantages using novel pathways offer.
| + | We tried to expand on what we already did in our metabolic engineering project and tap the huge advantages using novel pathways offer. |
| − | We tried to enable a novel pathway by engineering an enzyme to catalyze a new reaction which corresponds to the 4th level of metabolic engineering.
| + | We tried to enable a novel pathway by engineering an enzyme to catalyze a new reaction which corresponds to the 4th level of metabolic engineering. |
| | </p> | | </p> |
| | <figure style="float: right; width: 400px"> | | <figure style="float: right; width: 400px"> |
| − | <img style="float:right; width:100%;" src="https://ars.els-cdn.com/content/image/1-s2.0-S1367593116302071-gr1.jpg" alt="text" width="70%">
| + | <img style="float:right; width:100%;" src="https://ars.els-cdn.com/content/image/1-s2.0-S1367593116302071-gr1.jpg" alt="text" width="70%"> |
| − | <figcaption style="float: right;"><b>Figure 1:</b> Different levels of metabolic engineering and their respective biochemical space.<a href="https://www.sciencedirect.com/science/article/pii/S1367593116302071"><abbr title ="Tobias J Erb, Patrik R Jones, Arren Bar-Even, Synthetic metabolism: metabolic engineering meets enzyme design, Current Opinion in Chemical Biology (2017) 56-62" >(Erb <i>et al.</i>2017)</abbr></a></figcaption>
| + | <figcaption style="float: right;"><b>Figure 1:</b> Different levels of metabolic engineering and their respective biochemical space.<a href="https://www.sciencedirect.com/science/article/pii/S1367593116302071"><abbr title ="Tobias J Erb, Patrik R Jones, Arren Bar-Even, Synthetic metabolism: metabolic engineering meets enzyme design, Current Opinion in Chemical Biology (2017) 56-62" >(Erb <i>et al.</i>2017)</abbr></a></figcaption> |
| | </figure> | | </figure> |
| | <h3>Design of the binding pocket</h3> | | <h3>Design of the binding pocket</h3> |
| | <p> | | <p> |
| − | The pathway we chose for our metabolic engineering efforts was - in our opinion - the best pathway that has been explored previously.
| + | The pathway we chose for our metabolic engineering efforts was - in our opinion - the best pathway that has been explored previously. |
| − | But there are many more possible theoretical
| + | But there are much more possible theoretical |
| − | <a href="https://link.springer.com/article/10.1007/s00253-013-4802-4"><abbr title ="Kris Niño G. Valdehuesa, Huaiwei Liu, Grace M. Nisola, Wook-Jin Chung, Seung Hwan Lee, Si Jae Park, Recent advances in the metabolic engineering of microorganisms for the production of 3-hydroxypropionic acid as C3 platform chemical, Applied Microbiology and Biotechnology (2013), Volume 97, Issue 8, pp 3309–332" >(Valdehuesa <i>et al.</i>2013)</abbr></a>
| + | <a href="https://link.springer.com/article/10.1007/s00253-013-4802-4"><abbr title ="Kris Niño G. Valdehuesa, Huaiwei Liu, Grace M. Nisola, Wook-Jin Chung, Seung Hwan Lee, Si Jae Park, Recent advances in the metabolic engineering of microorganisms for the production of 3-hydroxypropionic acid as C3 platform chemical, Applied Microbiology and Biotechnology (2013), Volume 97, Issue 8, pp 3309–332" >(Valdehuesa <i>et al.</i>2013)</abbr></a> |
| − | pathways with remarkable properties that have not been explored yet.
| + | pathways with remarkable properties that have not been explored yet. |
| − | The reason is most of the times that it involves one or more reactions with no known enzyme to catalyze them.
| + | The reason is most of the times that it involves one or more reactions with no known enzyme to catalyze them. |
| − | One theoretical pathway is from a free energy standpoint much more favorable, but there is one step without a known enzyme to catalyze it. We performed an intense literature research and decided to try to build an enzyme capable of decarboxylating malate to <dfn data-info="3-hydroxypropionic acid">3HPA</dfn>. Even with <i> Vibrio Natriegens </i> if we would try to engineer an enzyme to catalyze the wanted reaction using random mutagenesis it would take ages if it succeeds at all. That is why we decided to use a combination of <i>in silico</i> as well as wet lab methods to boost our chances of succeeding.
| + | One theoretical pathway is from a free energy standpoint much more favorable, but there is one step without a known enzyme to catalyze it. We performed an intense literature research and decided to try to build an enzyme capable of decarboxylating malate to <dfn data-info="3-hydroxypropionic acid">3HPA</dfn>. Even with <i> Vibrio Natriegens </i> if we would try to engineer an enzyme to catalyze the wanted reaction using random mutagenesis it would take ages if it succeeds at all. That is why we decided to use a combination of <i>in silico</i> as well as wet lab methods to boost our chances of succeeding. |
| | </p> | | </p> |
| | <br> | | <br> |
| | <figure style="width: 600px; float: right;"> | | <figure style="width: 600px; float: right;"> |
| − | <img width="100%" src="https://static.igem.org/mediawiki/2018/2/25/T--Marburg--pathways_notfinal.png" alt="text">
| + | <img width="100%" src="https://static.igem.org/mediawiki/2018/2/25/T--Marburg--pathways_notfinal.png" alt="text"> |
| − | <figcaption><b>Figure 2:</b> Comparison between established and theoretical pathway using free energy. Reaction with no enzyme to catalyze marked with a green star.</figcaption>
| + | <figcaption><b>Figure 2:</b> Comparison between established and theoretical pathway using free energy. Reaction with no enzyme to catalyze marked with a green star.</figcaption> |
| | </figure> | | </figure> |
| | <br> | | <br> |
| | <p> | | <p> |
| − | The pathway that we want to enable is displayed in Figure [2]. The step that has to be catalyzed is the decarboxylation reaction of malate to our final product, <dfn data-info="3-hydroxypropionic acid">3HPA</dfn>. As a start, we investigated the enzyme family of Carboxy-lyases (EC Number 4.1.1). We - in accordance to literature - were not able to find an enzyme that can catalyze this reaction.
| + | The pathway that we want to enable is displayed in Figure 2. The step that has to be catalyzed is the decarboxylation reaction of malate to our final product, <dfn data-info="3-hydroxypropionic acid">3HPA</dfn>. As a start, we investigated the enzyme family of Carboxy-lyases (EC Number 4.1.1). We - in accordance with literature - were not able to find an enzyme that can catalyze this reaction. |
| − | However, there were some that we thought could help us to develope a binding pocket capable of decarboxylating malate.
| + | However, there were some that we thought could help us to develop a binding pocket capable of decarboxylating malate. |
| | <br> | | <br> |
| − | One of those was acetolactate decarboxylase (<dfn data-info="Acetolactate Decarboxylase">ALD</dfn> 4.1.1.5).
| + | One of those was acetolactate decarboxylase (<dfn data-info="Acetolactate Decarboxylase">ALD</dfn> 4.1.1.5). |
| − | The important difference from its natural substrate to malate being that there is a carboxy group in β position (see Figure X).
| + | The important difference from its natural substrate to malate being that there is a carboxy group in β position (see Figure 4). |
| − | In this enzyme with the help of this carboxy group a double bond is formed to an intermediate product (see Figure X) that we cannot form with malate as substrate. But the zinc cation that is used as a cofactor should be able to bind to malate in a similar way as it does with the natural substrate.
| + | In this enzyme with the help of this carboxy group a double bond is formed to an intermediate product (see Figure 4) that we cannot form with malate as substrate. But the zinc cation that is used as a cofactor should be able to bind to malate in a similar way as it does with the natural substrate. |
| − | This could be a promising starting point, because we would be able to sustain a specific conformation of the substrate and alter the electronic structure of the substrate at the same time.
| + | This could be a promising starting point because we would be able to sustain a specific conformation of the substrate and alter the electronic structure of the substrate at the same time. |
| − | The complex of malate inside the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> binding pocket is shown in Figure [3].
| + | The complex of malate inside the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> binding pocket is shown in Figure 3. |
| − | The zinc cofactor is bound by three histidines and the zinc binds to the malate (or the substrate analogue in the real crystal structure) with three interactions.
| + | The zinc cofactor is bound by three histidines and the zinc binds to the malate (or the substrate analog in the real crystal structure) with three interactions. |
| − | There is another important residue close to this complex, Arg145.
| + | There is another important residue close to this complex, Arg145. |
| − | After the double bond and with it the intermediate product is formed, this residue is protonating the intermediate to form the product.
| + | After the double bond and with it the intermediate product is formed, this residue is protonating the intermediate to form the product. |
| | </p> | | </p> |
| | <figure style="width: 500px; float: left;"> | | <figure style="width: 500px; float: left;"> |
| − | <img style="width: 100%" src="https://static.igem.org/mediawiki/2018/thumb/2/2a/T--Marburg--ald_binding_pocket_with_malate.png/1600px-T--Marburg--ald_binding_pocket_with_malate.png" alt="text">
| + | <img style="width: 100%" src="https://static.igem.org/mediawiki/2018/thumb/2/2a/T--Marburg--ald_binding_pocket_with_malate.png/1600px-T--Marburg--ald_binding_pocket_with_malate.png" alt="text"> |
| − | <figcaption><b>Figure 3:</b> Complex of Malate with the Zn Cofactor inside the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> binding pocket [PDB 4BT3]. .</figcaption>
| + | <figcaption><b>Figure 3:</b> Complex of Malate with the Zn Cofactor inside the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> binding pocket [PDB 4BT3]. .</figcaption> |
| | </figure> | | </figure> |
| | <br> | | <br> |
| | <p> | | <p> |
| − | Another enzyme with an enzyme mechanism that might help us find a way to catalyze this reaction is Orotidine 5'-phosphate decarboxylase (<dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> 4.1.1.23).
| + | Another enzyme with an enzyme mechanism that might help us find a way to catalyze this reaction is Orotidine 5'-phosphate decarboxylase (<dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> 4.1.1.23). |
| − | The reaction <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> catalyzes compared to the reaction of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> and the one we want to catalyze is shown in Figure X.
| + | The reaction <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> catalyzes compared to the reaction of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> and the one we want to catalyze is shown in Figure 4. |
| − | <br>
| + | <br> |
| − | <figure style="width: 400px; float: right;">
| + | <figure style="width: 400px; float: right;"> |
| − | <img style="display:block; margin:0 auto 0 auto; width:400px;" src="https://static.igem.org/mediawiki/2018/9/90/T--Marburg--vergleich_substrates_reactions_notfinal.png" alt="text" width="100%">
| + | <img style="display:block; margin:0 auto 0 auto; width:400px;" src="https://static.igem.org/mediawiki/2018/9/90/T--Marburg--vergleich_substrates_reactions_notfinal.png" alt="text" width="100%"> |
| − | <figcaption><b>Figure X:</b> Comparison between the reaction catalyzed by <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> , <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> and the reaction that we want to catalyze. .</figcaption>
| + | <figcaption><b>Figure 4:</b> Comparison between the reaction catalyzed by <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> , <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> and the reaction that we want to catalyze. .</figcaption> |
| | </figure> | | </figure> |
| | <br> | | <br> |
| Line 327: |
Line 326: |
| | The reaction is split into two steps, first the decarboxylation and a simultaneous salt bridge between a lysine and the resulting carbanion. | | The reaction is split into two steps, first the decarboxylation and a simultaneous salt bridge between a lysine and the resulting carbanion. |
| | After this, the lysine protonates the carbanion to yield the final product. | | After this, the lysine protonates the carbanion to yield the final product. |
| − | The most important difference between the substrate of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> and malate is, that the carbanion stabilized (Figure X) in the former one whilst there is nearly no stabilization in the latter one. | + | The most important difference between the substrate of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> and malate is, that the carbanion stabilized (Figure 4) in the former one whilst there is nearly no stabilization in the latter one. |
| | The idea of our enzyme design is that we use the direct decarboxylation of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> and try to stabilize the carbanion with the cofactor of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn>. | | The idea of our enzyme design is that we use the direct decarboxylation of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> and try to stabilize the carbanion with the cofactor of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn>. |
| − | With this plan, we need to engineer the pocket in a way that a lysine side chain can get to the carbon atom of malate that shall be protonated. | + | With this plan, we need to engineer the pocket in a way that a lysine side chain can get to the C2-carbon of malate that shall be protonated. |
| | Even if we can place a lysine near to the substrate, the reaction is not automatically working since the transition state energy is probably too high. | | Even if we can place a lysine near to the substrate, the reaction is not automatically working since the transition state energy is probably too high. |
| | We need to calculate the transition state energy of our engineered systems and compare it to the literature to estimate the feasibility of our reaction. | | We need to calculate the transition state energy of our engineered systems and compare it to the literature to estimate the feasibility of our reaction. |
| | If the reaction is not feasible with a single lysine in the binding pocket, we need to alter the binding pocket to lower the transition state energy. | | If the reaction is not feasible with a single lysine in the binding pocket, we need to alter the binding pocket to lower the transition state energy. |
| − | After that we need to mutate an enzyme to resemble the binding pocket. | + | After that, we need to mutate an enzyme to resemble the binding pocket. |
| | <br> | | <br> |
| | <br> | | <br> |
| − | As a start point we looked at all single point mutations to lysine that can be done where the protonated nitrogen of lysine has the possibility to get as close to the malate, since this is required for reprotonation. All Mutations investigated are displayed in table 1.
| + | As a start point, we looked at all single point mutations to lysine that can be done where the protonated nitrogen of lysine has the possibility to get as close to the malate since this is required for reprotonation. All Mutations investigated are displayed in table 1. |
| − | In the natural binding pocket of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> there is an arginine residue that is used for reprotonation of the natural substrate.
| + | In the natural binding pocket of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> there is an arginine residue that is used for reprotonation of the natural substrate. |
| − | Because of the possibility that this disturbs the lysine we mutated that in each binding pocket where it is not already mutated to a glycine.
| + | Because of the possibility that this disturbs the lysine we mutated that in each binding pocket where it is not already mutated to a glycine. |
| | | | |
| | </p> | | </p> |
| | | | |
| | <table align="center" style="width:100%";> | | <table align="center" style="width:100%";> |
| − | <caption> <b>Table 1:</b> All point mutations to <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> (PDB 4BT3) that have been investigated. The aminoacids are displayed as single character with the residue number in between. All mutations besides R145K also include R145G. </caption>
| + | <caption> <b>Table 1:</b> All point mutations to <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> (PDB 4BT3) that have been investigated. The aminoacids are displayed as single character with the residue number in between. All mutations besides R145K also include R145G. </caption> |
| | <tr> | | <tr> |
| | <th>L34K</th> | | <th>L34K</th> |
| Line 360: |
Line 359: |
| | </table> | | </table> |
| | | | |
| − | <div class="skipTarget" skipname="Enzyme Design"></div>
| + | <div class="skipTarget" skipname="Enzyme Design"></div> |
| | | | |
| | <h3><i>in silico</i> enzyme design</h3> | | <h3><i>in silico</i> enzyme design</h3> |
| | <p> | | <p> |
| − | We developed a double modeling approach to investigate the whole system to the best of our ability. First, we try to investigate the reaction using quantum mechanical calculations (<dfn data-info="Quantum Mechanics">QM</dfn>) and model the best possible system.
| + | We developed a double modeling approach to investigate the whole system to the best of our ability. First, we try to investigate the reaction using quantum mechanical calculations (<dfn data-info="Quantum Mechanics">QM</dfn>) and model the best possible system. |
| − | Then we control how well this system is retained if we make certain mutations in the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> with the help of molecular dynamic (<dfn data-info="Molecular Dynamics">MD</dfn> ) Simulations.
| + | Then we control how well this system is retained if we make certain mutations in the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> with the help of molecular dynamic (<dfn data-info="Molecular Dynamics">MD</dfn> ) Simulations. |
| | </p> | | </p> |
| | | | |
| Line 372: |
Line 371: |
| | <h3>Quantum mechanics calculations </h3> | | <h3>Quantum mechanics calculations </h3> |
| | <p> | | <p> |
| − | First, we wanted to model the reaction that we try to catalyze using <dfn data-info="Quantum Mechanics">QM</dfn> level calculations. We used density functional theory based method b3-lyp with different basis sets. With these calculations, we tried to get a further insight into the reaction coordinate and all corresponding energies, most importantly the activation energy of the reaction.
| + | First, we wanted to model the reaction that we try to catalyze using <dfn data-info="Quantum Mechanics">QM</dfn> level calculations. We used density functional theory based method b3-lyp with different basis sets. With these calculations, we tried to get a further insight into the reaction coordinate and all corresponding energies, most importantly the activation energy of the reaction. |
| | The activation energy can be calculated as the difference between the energy of the educts and the transition state energy. | | The activation energy can be calculated as the difference between the energy of the educts and the transition state energy. |
| | The activation energy is crucial for the activity of the final enzyme and high activation energies correspond to low or no activity of the final enzyme. | | The activation energy is crucial for the activity of the final enzyme and high activation energies correspond to low or no activity of the final enzyme. |
| Line 387: |
Line 386: |
| | | | |
| | <h4>Building of binding pockets</h4> | | <h4>Building of binding pockets</h4> |
| − | <p> As a start point for a structure we used the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> crystal structure of | + | <p> As a starting point we used the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> crystal structure of |
| − | <a href="https://pubs.acs.org/doi/abs/10.1021/cb400429h"><abbr title ="Victoria A. Marlow, Dean Rea, Shabir Najmudin, Martin Wills, and Vilmos Fülöp, PStructure and Mechanism of Acetolactate Decarboxylase, ACS Chem. Biol., 2013, 8 (10), pp 2339–2344" >(Marlow <i>et al.</i>2013)</abbr></a>
| + | <a href="https://pubs.acs.org/doi/abs/10.1021/cb400429h"><abbr title ="Victoria A. Marlow, Dean Rea, Shabir Najmudin, Martin Wills, and Vilmos Fülöp, PStructure and Mechanism of Acetolactate Decarboxylase, ACS Chem. Biol., 2013, 8 (10), pp 2339–2344" >(Marlow <i>et al.</i>2013)</abbr></a> |
| − | (PDB CODE 4BT3).
| + | (PDB CODE 4BT3). |
| − | From that, we extracted the position of the three zinc coordinating histidines, the zinc cation, and the substrate and changed the substrate to malate. | + | From that, we extracted the position of the three zinc coordinating histidines, the zinc cation, and the substrate and changed the substrate analog ((2R,3R)-2,3-Dihydroxy-2-methylbutanoic acid) to malate. |
| − | We then built multiple differently binding pockets that are displayed in Table [2]. | + | We then built multiple different binding pockets that are displayed in Table [2]. |
| | </p> | | </p> |
| | <table style="width:100%" cellspacing="5" border="1"> | | <table style="width:100%" cellspacing="5" border="1"> |
| − | <caption> <b>Table 2:</b> The differently setup systems with the changes made and names given.</caption>
| + | <caption> <b>Table 2:</b> The differently setup systems with the changes made and names given.</caption> |
| − | <tr>
| + | <tr> |
| − | <th>Kind of Binding Pocket</th>
| + | <th>Type of Binding Pocket</th> |
| − | <th>Abbreviation used in Graphs</th>
| + | <th>Abbreviation used in Graphs</th> |
| − | <th>Residues/Molecules involved</th>
| + | <th>Residues/Molecules involved</th> |
| − | </tr>
| + | </tr> |
| − | <tr>
| + | <tr> |
| − | <td>Binding Pocket without Cofactor</td>
| + | <td>Binding Pocket without Cofactor</td> |
| − | <td>Complex</td>
| + | <td>Complex</td> |
| − | <td>Lysine, Substrate</td>
| + | <td>Lysine, Substrate</td> |
| − | </tr>
| + | </tr> |
| − | <tr>
| + | <tr> |
| − | <td>Binding Pocket</td>
| + | <td>Binding Pocket</td> |
| − | <td>Nor_1</td>
| + | <td>Nor_1</td> |
| − | <td>Three Histidines, Zinc cation, Lysine, Glutamate, Substrate</td>
| + | <td>Three Histidines, Zinc cation, Lysine, Glutamate, Substrate</td> |
| − | </tr>
| + | </tr> |
| − | <tr>
| + | <tr> |
| − | <td>Binding Pocket pre-optimized</td>
| + | <td>Binding Pocket pre-optimized</td> |
| − | <td>Nor_2</td>
| + | <td>Nor_2</td> |
| − | <td>Three Histidines, Zinc cation, Lysine, Glutamate, Substrate</td>
| + | <td>Three Histidines, Zinc cation, Lysine, Glutamate, Substrate</td> |
| − | </tr>
| + | </tr> |
| − | <tr>
| + | <tr> |
| − | <td>Minimal Binding Pocket</td>
| + | <td>Minimal Binding Pocket</td> |
| − | <td>Min</td>
| + | <td>Min</td> |
| − | <td>Three Histidines, Zinc cation, Lysine, Substrate</td>
| + | <td>Three Histidines, Zinc cation, Lysine, Substrate</td> |
| − | </tr>
| + | </tr> |
| − | <tr>
| + | <tr> |
| − | <td>Binding Pocket with two lysine residues</td>
| + | <td>Binding Pocket with two lysine residues</td> |
| − | <td>2ly</td>
| + | <td>2ly</td> |
| − | <td>Three Histidines, Zinc cation, Lysine, Lysine, Substrate</td>
| + | <td>Three Histidines, Zinc cation, Lysine, Lysine, Substrate</td> |
| − | </tr>
| + | </tr> |
| | </table> | | </table> |
| | <p> | | <p> |
| − | Because we wanted to resemble the binding pocket to the best of our knowledge we made a system where we kept Glu 253 and added a lysine close to the carbon that it shall protonate.
| + | Because we wanted to resemble the binding pocket to the best of our knowledge we made a system in which we kept Glu253 and added a lysine close to the C2-carbon that it should protonate. |
| | Later we used a pre-optimized structure of previous calculations and used this pre-optimized structure to build a minimal binding pocket where we removed Glu. | | Later we used a pre-optimized structure of previous calculations and used this pre-optimized structure to build a minimal binding pocket where we removed Glu. |
| | We also made one system with two lysine residues. | | We also made one system with two lysine residues. |
| | We will go into detail on why and how we designed this system when we explain the hypothesis. | | We will go into detail on why and how we designed this system when we explain the hypothesis. |
| − | As a start point we also used a structure of just malate and lysine. | + | As a start point, we also used a structure of malate and lysine, referred to as complex in Table 2. |
| − | The final systems are displayed in Figure BLA. | + | The final systems are displayed in Figure 5. |
| | | | |
| | <figure> | | <figure> |
| − | <div class="imageContainer2x2">
| + | <div class="imageContainer2x2"> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/thumb/0/03/T--Marburg--pre_converged_1.png/1600px-T--Marburg--pre_converged_1.png">a</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/thumb/0/03/T--Marburg--pre_converged_1.png/1600px-T--Marburg--pre_converged_1.png">a</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/thumb/1/11/T--Marburg--post_converged_binding_pocket.png/1600px-T--Marburg--post_converged_binding_pocket.png">b</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/thumb/1/11/T--Marburg--post_converged_binding_pocket.png/1600px-T--Marburg--post_converged_binding_pocket.png">b</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/thumb/3/38/T--Marburg--minimal_binding_pocket.png/1600px-T--Marburg--minimal_binding_pocket.png">c</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/thumb/3/38/T--Marburg--minimal_binding_pocket.png/1600px-T--Marburg--minimal_binding_pocket.png">c</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/thumb/4/48/T--Marburg--binding_pocket_2lys.png/1600px-T--Marburg--binding_pocket_2lys.png">d</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/thumb/4/48/T--Marburg--binding_pocket_2lys.png/1600px-T--Marburg--binding_pocket_2lys.png">d</div> |
| − | </div>
| + | </div> |
| − | <figcaption>
| + | <figcaption> |
| − | Figure X : Displayed of the different binding pockets. a) pre converged full binding pocket b) post converged full binding pocket c) minimal binding pocket d) binding pocket with two lysine residues
| + | Figure 5 : Displayed of the different binding pockets. a) pre converged full binding pocket b) post converged full binding pocket c) minimal binding pocket d) binding pocket with two lysine residues |
| − | </figcaption>
| + | </figcaption> |
| − | </figure>
| + | </figure> |
| − | To evaluate the transition state energy and the overall energy profile of the reaction an investigation of the reaction path is necessary.
| + | To evaluate the transition state energy and the overall energy profile of the reaction an investigation of the reaction path is necessary. |
| − | Since we want to investigate a decarboxylation reaction, which is a c-c bond break, we chose to set up multiple calculations at different c-c bond lengths to evaluate the energy necessary to split that bond.
| + | Since we want to investigate a decarboxylation reaction, which is a c-c bond break, we chose to set up multiple calculations at different c-c bond lengths to evaluate the energy necessary to split that bond. |
| − | This way we sample the reaction coordinate of the systems.
| + | This way we sample the reaction coordinate of the systems. |
| − | In 20 calculations we added from 0 to 2 Angstroem to the c-c bond length in 0.1 Angstroem steps.
| + | We screened the bond length in 0.1 Angstroem steps and added between 0 and 2 Angstroem to the starting c-c distance, this results in 20 calculations per screen. |
| − | With this method, we performed calculations for the starting system (no added c-c distance), the transition state (varying c-c distances depending on the system) and the end product (high c-c distances).
| + | With this method, we performed calculations for the starting system (no added c-c distance), the transition state (varying c-c distances depending on the system) and the final product (high c-c distances). |
| | </p> | | </p> |
| | | | |
| Line 456: |
Line 455: |
| | <p> | | <p> |
| | | | |
| − | <figure style="width: 50%; float: right;">
| + | <figure style="width: 50%; float: right;"> |
| − | <div class="imageContainer2x2">
| + | <div class="imageContainer2x2"> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/0/08/T--Marburg--complex_ccpvdz_1.png">A</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/0/08/T--Marburg--complex_ccpvdz_1.png">a</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/6/6b/T--Marburg--nor1_631g.png">B</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/6/6b/T--Marburg--nor1_631g.png">b</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/7/7d/T--Marburg--nor2_ccpvdz.png">C</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/7/7d/T--Marburg--nor2_ccpvdz.png">c</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/2/28/T--Marburg--min_631g_1.png">D</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/2/28/T--Marburg--min_631g_1.png">d</div> |
| | </div> | | </div> |
| − | <figcaption><b>Figure 5:</b> Energy Profiles of C-C bond lengthening</figcaption>
| + | <figcaption><b>Figure 6:</b> Energy Profiles of C-C bond lengthening</figcaption> |
| | </figure> | | </figure> |
| − | The results of the calculations can be seen in Figure [5].
| + | The results of the calculations can be seen in Figure 6. |
| | <br> | | <br> |
| − | Just for the complex the activation barrier is 59.04 kcal/mol. | + | The calculated activation barrier for the complex (cc-pVDZ basis set) is 59.04 kcal/mol [Figure 6a]. |
| | This is far over the free energy barrier of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> in the literature (15.54 kcal/mol <a href="https://pubs.acs.org/doi/abs/10.1021/jp074858n"><abbr title ="Courtney L. Stanton, I-Feng W. Kuo, Christopher J. Mundy, Teodoro Laino, and K. N. Houk, QM/MM Metadynamics Study of the Direct Decarboxylation Mechanism for Orotidine-5‘-monophosphate Decarboxylase Using Two Different QM Regions: Acceleration Too Small To Explain Rate of Enzyme Catalysis, J. Phys. Chem. B, <b>2007 </b>, 111 (43), pp 12573–12581" >(Courtney <i>et al.</i>2007)</abbr></a>). | | This is far over the free energy barrier of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> in the literature (15.54 kcal/mol <a href="https://pubs.acs.org/doi/abs/10.1021/jp074858n"><abbr title ="Courtney L. Stanton, I-Feng W. Kuo, Christopher J. Mundy, Teodoro Laino, and K. N. Houk, QM/MM Metadynamics Study of the Direct Decarboxylation Mechanism for Orotidine-5‘-monophosphate Decarboxylase Using Two Different QM Regions: Acceleration Too Small To Explain Rate of Enzyme Catalysis, J. Phys. Chem. B, <b>2007 </b>, 111 (43), pp 12573–12581" >(Courtney <i>et al.</i>2007)</abbr></a>). |
| − | If we now take a look at the activation barrier with the normal binding pocket with the 6-31g* basis set (53.48). | + | If we now take a look at the activation barrier calculated for the nor1 system with the 6-31g* basis set (53.48 kcal/mol) [Figure 6c], then this is not what we expected or wanted to see. |
| − | This is not what we expected or wanted to see since this means the addition of the zinc complex that shall stabilize the carbanion only benefits the reaction with around 6 kcal/mol.
| + | The addition of the zinc complex that shall stabilize the carbanion only benefits the reaction with around 6 kcal/mol. |
| | The transition states that we calculate are confirmed using frequency calculations. | | The transition states that we calculate are confirmed using frequency calculations. |
| − | If there is just one negative frequency calculated for a optimized structure, this means that it is a transition state. | + | If there is just one negative frequency calculated for an optimized structure, this means that it is a transition state. |
| | The frequency can then be animated, to show the movement that is enforced by it. | | The frequency can then be animated, to show the movement that is enforced by it. |
| − | An animation of the negative frequency of the minimal binding pocket transition state can be seen in Figure 6. | + | An animation of the negative frequency of the minimal binding pocket transition state can be seen in Figure 7. |
| − | <figure style="width: 600px; float: left;">
| + | <figure style="width: 600px; float: left;"> |
| − | <img src="https://static.igem.org/mediawiki/2018/9/93/T--Marburg--ts_frequency_animation.png" alt="text" width="100%" height="100%"> <figcaption><b>Figure 6:</b> Animation of negative frequency of the transition state of the normal binding pocket.</figcaption>
| + | <img src="https://static.igem.org/mediawiki/2018/9/93/T--Marburg--ts_frequency_animation.png" alt="text" width="100%" height="100%"> <figcaption><b>Figure 7:</b> Animation of negative frequency of the transition state of the normal binding pocket.</figcaption> |
| | </figure> | | </figure> |
| − | All of the calculations that we do take quite a long time and to reduce the computational cost we minimized the starting structure further before we tried to calculate the normal binding pocket with a larger basis set and the minimal binding pocket.
| + | Since all electronic structure calculations conducted in this study are computationally quite demanding, starting structures were pregenerated by structure optimizations on a low level of theory. The optimized structures are then used to build the starting structures for high-level calculations on the normal binding pocket and the minimal binding pocket. |
| − | As minimizations are heavily dependent on the starting structure, we got different results with these structures than we expected. | + | As geometric optimizations are heavily dependent on the starting structure, we got different results with these structures than we got previously. |
| − | For both calculations, the activation barrier was approximately 7 kcal/mol less than in the previous calculations. | + | The Nor_2 (cc-pVDZ basis set) [Figure 6b] and the min (6-31g* basis set) [Figure 6d] electronic structure calculations were set up using a pre converged structure. |
| | + | For these two calculations, the activation barrier was approximately 7 kcal/mol less than in the previous calculations. |
| | This was due to the fact that the lysine was not in the intended place at the end of the minimization. | | This was due to the fact that the lysine was not in the intended place at the end of the minimization. |
| − | Rather than building the salt bridge with the carbanion, it was interacting with both carboxy functions, which was lowering the activation energy. | + | Rather than building the salt bridge with the carbanion, it was interacting with both carboxy moieties, which was lowering the activation energy. |
| | <br> | | <br> |
| | <br> | | <br> |
| − | This was boosting our hope that if we introduce the correct changes to the binding pocket, we could change the electronic structure of the ligand in a way that reduces the activation barrier enough. | + | This was boosting our hope that if we introduce the correct changes to the binding pocket, we could change the electronic structure of malate in a way to sufficiently reduce the activation barrier. |
| − | To use this knowledge we set up another system with two lysine residues, one interacting with both carboxy functions and one that shall protonate the carbanion in the hopes of lowering the activation energy even further. | + | To use this knowledge we set up another system with two lysine residues, one interacting with both carboxy moieties (lys1) and one that shall protonate the C2-carbon (lys2). |
| − | Our expectations were subverted multiple times.
| + | The activation barrier (2lys 631g*) was slightly lower than before (~ 43 kcal/mol), but lys2 was not reprotonating the carbanion but interacting with the hydroxy moiety. |
| − | The activation barrier was slightly lower than before (~ 43 kcal/mol), but the second lysine was again not reprotonating the carbanion but interacting with the hydroxy group. | + | Even more interesting was the fact that the carbanion formed a covalent bond with the zinc cation with ca. 10 kcal/mol less energy than the transition state. |
| − | Far more interesting was the fact that the carbanion formed a covalent bond with the zinc cation with ca. 10 kcal/mol less energy than the transition state. | + | |
| | The optimized structure with the covalent bond between the zinc cation and the malate is displayed in Figure 8. | | The optimized structure with the covalent bond between the zinc cation and the malate is displayed in Figure 8. |
| − | <figure style="width: 400px; float: right;" >
| + | <figure style="width: 400px; float: right;" > |
| − | <img src="https://static.igem.org/mediawiki/2018/d/d4/T--Marburg--malate_bound_to_zinc.jpg" alt="text" width="100%">
| + | <img src="https://static.igem.org/mediawiki/2018/d/d4/T--Marburg--malate_bound_to_zinc.jpg" alt="text" width="100%"> |
| − | <figcaption><b>Figure 8:</b> Intermediate state of the reaction with covalent bond between zinc cation and malate.</figcaption>
| + | <figcaption><b>Figure 8:</b> Intermediate state of the reaction with covalent bond between zinc cation and malate.</figcaption> |
| | </figure> | | </figure> |
| − | This adds a new way that the reaction can take place that can be used as an alternative approach without need of direct reprotonation.
| + | This adds a new way at which the reaction can take place that can be used as an alternative approach without the need of direct reprotonation. |
| | </p> | | </p> |
| | | | |
| Line 502: |
Line 501: |
| | <h3>Molecular Dynamic (<dfn data-info="Molecular Dynamics">MD</dfn> ) Simulations </h3> | | <h3>Molecular Dynamic (<dfn data-info="Molecular Dynamics">MD</dfn> ) Simulations </h3> |
| | <p> | | <p> |
| − | To evaluate how well the system that we tried to engineer in the <dfn data-info="Quantum Mechanics">QM</dfn> calculations is represented in the complete enzyme systems we performed <dfn data-info="Molecular Dynamics">MD</dfn> simulations with <i>in silico</i> mutated enzyme versions.
| + | To evaluate how well the system that we tried to engineer in the <dfn data-info="Quantum Mechanics">QM</dfn> calculations is represented in the complete enzyme systems we performed <dfn data-info="Molecular Dynamics">MD</dfn> simulations with <i>in silico</i> mutated enzyme versions. |
| − | We performed 200 ns <dfn data-info="Molecular Dynamics">MD</dfn> Simulations of all previously mentioned mutated systems as well as the wild-type enzyme with 3 replicas each.
| + | We performed 200 ns <dfn data-info="Molecular Dynamics">MD</dfn> Simulations of all previously mentioned mutated systems as well as the wild-type enzyme with 3 replicas each. |
| − | With the help of these <dfn data-info="Molecular Dynamics">MD</dfn> simulations, we are able to evaluate how well the different mutations resemble the system that we designed for the <dfn data-info="Quantum Mechanics">QM</dfn> calculations.</p>
| + | With the help of these <dfn data-info="Molecular Dynamics">MD</dfn> simulations, we are able to evaluate how well the different mutations resemble the system that we designed for the <dfn data-info="Quantum Mechanics">QM</dfn> calculations.</p> |
| | <h4>Setup of <dfn data-info="Molecular Dynamics">MD</dfn> Simulations</h4> | | <h4>Setup of <dfn data-info="Molecular Dynamics">MD</dfn> Simulations</h4> |
| | <p> | | <p> |
| − | For the <dfn data-info="Molecular Dynamics">MD</dfn> simulations, we used the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> crystal structure of [Reference] (PDB CODE 4BT3).
| + | For the <dfn data-info="Molecular Dynamics">MD</dfn> simulations, we used the <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> crystal structure of [Reference] (PDB CODE 4BT3). |
| | We changed the substrate to malate and made the corresponding mutations for each system. | | We changed the substrate to malate and made the corresponding mutations for each system. |
| | Then we capped the termini of the enzyme, checked for missing residues and protonated all residues according to pH 7.0. | | Then we capped the termini of the enzyme, checked for missing residues and protonated all residues according to pH 7.0. |
| − | We visually inspected the residues close to the binding pocket to make sure that all protonation states seem correct.</p> | + | We visually inspected the residues close to the binding pocket to make sure that all protonation states are correctly assigned.</p> |
| | <h4>Results of <dfn data-info="Molecular Dynamics">MD</dfn> Simulations</h4> | | <h4>Results of <dfn data-info="Molecular Dynamics">MD</dfn> Simulations</h4> |
| | | | |
| | <p> | | <p> |
| − | To evaluate the different systems that we did <dfn data-info="Molecular Dynamics">MD</dfn> Simulations on we developed a mechanical descriptor.
| + | We previously established the mutations we introduced <i>in silico</i> (Table 1) to <dfn data-info="Acetolactate Decarboxylase">ALD</dfn>. |
| − | The most important thing in our case was the distance between the lysine side chain and the malate to see how well each variation resembles the structure that we assumed for the <dfn data-info="Quantum Mechanics">QM</dfn> calculations. | + | For evaluating how well the mutants resemble the binding pocket developed with the help of the <dfn data-info="Quantum Mechanics">QM</dfn> we developed a mechanical descriptor. |
| − | We measured the distance between the lysine sidechains nitrogen atom and the carbon atom that shall be protonated of the malate for every frame of every <dfn data-info="Molecular Dynamics">MD</dfn> Simulation. | + | The distance between the binding pockets lysine side chain and the malate is used. |
| − | An animation of one <dfn data-info="Molecular Dynamics">MD</dfn> Simulation with the distance highlighted is shown in Figure 9. | + | We measured the distance between the lysine sidechains nitrogen atom and the C2-carbon that should be protonated of the malate for every frame of every <dfn data-info="Molecular Dynamics">MD</dfn> Simulation. |
| − | <figure style="width: 800px; float: top;">
| + | This way we can evaluate if the lysine is close enough to the C2-Carbon for reprotonation as well as interaction with the carboxy moieties. |
| − | <img src="https://static.igem.org/mediawiki/2018/e/e1/T--Marburg--md_distance_1.gif" alt="text" width="100%">
| + | An animation of one <dfn data-info="Molecular Dynamics">MD</dfn> Simulation (L34K Replica 1) with the distance highlighted is shown in Figure 9. |
| − | <figcaption><b>Figure 9:</b> <dfn data-info="Molecular Dynamics">MD</dfn> Simulation of the L34K mutated system with the Malate-Lysine distance highlighted.</figcaption>
| + | <figure style="width: 800px; float: top;"> |
| | + | <img src="https://static.igem.org/mediawiki/2018/e/e1/T--Marburg--md_distance_1.gif" alt="text" width="100%"> |
| | + | <figcaption><b>Figure 9:</b> <dfn data-info="Molecular Dynamics">MD</dfn> Simulation of the L34K mutated system (replica 1) with the Malate-Lysine distance highlighted.</figcaption> |
| | </figure> | | </figure> |
| − | To display this data we chose to use normalized histograms.
| + | To display this data we chose to use normalized histograms. |
| | This way we can display the variation of this one variable. | | This way we can display the variation of this one variable. |
| − | That the Histograms are normalized means that we do not display the number of observations on the y-axis, but rather the density of observations in the bin. | + | Normalization means that we do not display the number of observations on the y-axis, but rather the density of observations in each bin. |
| − | If we multiply the bin width and the density of every bin and add the values up, the end value will be one. | + | The total area of the histogram is normalized to one. |
| | This helps to compare different histograms. | | This helps to compare different histograms. |
| − | We also colored each of the three trajectories that we simulated for each variation differently, so that we see if there is just one outlier or if all trajectories show the same behavior. | + | We also colored the three replicas that we simulated for each mutated enzyme differently, so that we can compare the different replicas. |
| | The data is displayed in Figure [10]. | | The data is displayed in Figure [10]. |
| | | | |
| | <figure> | | <figure> |
| − | <div class="imageContainer3x3">
| + | <div class="imageContainer3x3"> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/2/24/T--Marburg--gg_1dhist_L34K.png">a</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/2/24/T--Marburg--gg_1dhist_L34K.png">a</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/6/60/T--Marburg--gg_1dhist_L62K.png">b</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/6/60/T--Marburg--gg_1dhist_L62K.png">b</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/3/33/T--Marburg--gg_1dhist_R145K.png">c</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/3/33/T--Marburg--gg_1dhist_R145K.png">c</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/e/e2/T--Marburg--gg_1dhist_T58K.png">d</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/e/e2/T--Marburg--gg_1dhist_T58K.png">d</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/a/aa/T--Marburg--gg_1dhist_E65K.png">e</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/a/aa/T--Marburg--gg_1dhist_E65K.png">e</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/a/a2/T--Marburg--gg_1dhist_V147K.png">f</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/a/a2/T--Marburg--gg_1dhist_V147K.png">f</div> |
| | </div> | | </div> |
| − | <figcaption><b>Figure 10:</b> Histograms of all simulated trajectories. Histograms colored differently for each replica..</figcaption>
| + | <figcaption><b>Figure 10:</b> Histograms of all simulated trajectories. Histograms colored differently for each replica..</figcaption> |
| | </figure> | | </figure> |
| − | The first thing that stood out to us is that all the variations show mostly low distances and all replicas are in good agreement to each other. | + | The first thing that stood out to us is that all histograms show mostly low distance ( < 6 Angstroem) and most replicas (besides E65K_c [Figure 10e] and R145K_b [Figure 10c]) are in good agreement to each other. |
| − | We expected higher distances for some of them and more outliers than we were able to observe.
| + | |
| − | There are some outliers though, especially in the E65K and R145K variations we see that we sample distances that are far too large to allow for reprotonation.
| + | |
| | Apart from R145K and V147K all variants show distances under 4 Angstroem. | | Apart from R145K and V147K all variants show distances under 4 Angstroem. |
| | Especially L34K, T58K, and E65K have stable and low distances. | | Especially L34K, T58K, and E65K have stable and low distances. |
| Line 549: |
Line 548: |
| | This is very promising because it means that there is not only one potential position to introduce point mutations, which opens up further possibilities for enzyme design. | | This is very promising because it means that there is not only one potential position to introduce point mutations, which opens up further possibilities for enzyme design. |
| | To further distinguish between the different variations we decided to look at two dihedral angles of the lysine sidechain to evaluate its flexibility. | | To further distinguish between the different variations we decided to look at two dihedral angles of the lysine sidechain to evaluate its flexibility. |
| − | We do this with the help of two-dimensional histograms, which basically means that we overlap two histograms of different properties to see if and how they correlate. | + | To display this we used two-dimensional histograms. |
| − | We substituted the normal counts that would be on the z-axis with densities to help compare the different graphs. | + | The 2d histograms we developed contain all the information the 1d histograms had. |
| − | This means again that the integral over the complete 2d Histogram sums up to 1. | + | However, they do not "just" count how many instances of an investigation we have in a bin, but rather how many combinations of investigations we have in a small two-dimensional box. |
| − | The optimal candidate should show some flexibility so it is not entropically inhibited, but should also show consistent small distances. | + | This way we can not only investigate which distances each mutated enzyme has, but also pair this together with the corresponding dihedral angle(s). |
| − | The results are displayed in Figure [FIGURE] and [Figure]. | + | We also use normalized 2d-histograms, which means that the total area of the 2d-histogram is normalized to one. |
| | + | What we are looking for is a mutated enzyme which consistently shows low lysine-malate distances while simultaneously showing some flexibility in the dihedral angles. |
| | + | If the side chain of lysine shows too much flexibility (i.e. Figure 12d), the sidechains stability at the correct position is unfavorable. |
| | + | A ligand that shows too low flexibility (i.e. Figure 11b) the position is entropically unbeneficial. |
| | + | The results are displayed in Figure 11 and 12. |
| | | | |
| | | | |
| | </p> | | </p> |
| | <figure> | | <figure> |
| − | <div class="imageContainer3x3">
| + | <div class="imageContainer3x3"> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/c/c1/T--Marburg--gg_2dhist1_L34K.png">a</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/c/c1/T--Marburg--gg_2dhist1_L34K.png">a</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/2/20/T--Marburg--gg_2dhist1_L62K.png">b</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/2/20/T--Marburg--gg_2dhist1_L62K.png">b</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/9/9d/T--Marburg--gg_2dhist1_R145K.png">c</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/9/9d/T--Marburg--gg_2dhist1_R145K.png">c</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/a/a8/T--Marburg--gg_2dhist1_T58K.png">d</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/a/a8/T--Marburg--gg_2dhist1_T58K.png">d</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/f/f7/T--Marburg--gg_2dhist1_E65K.png">e</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/f/f7/T--Marburg--gg_2dhist1_E65K.png">e</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/8/80/T--Marburg--gg_2dhist1_V147K.png">f</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/8/80/T--Marburg--gg_2dhist1_V147K.png">f</div> |
| | </div> | | </div> |
| − | <figcaption><b>Figure X:</b> LOREM IPSUM</figcaption>
| + | <figcaption><b>Figure 11:</b>2D Histogram Lysine-Malate Distance vs Dihedral angle 1. a) L34K enzyme mutant b) R145K enzyme mutant c) V147K enzyme mutant d) L62K enzyme mutant e T58K enzyme mutant f) E65K enzyme mutant</figcaption> |
| | </figure> | | </figure> |
| | | | |
| | <figure> | | <figure> |
| − | <div class="imageContainer3x3">
| + | <div class="imageContainer3x3"> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/8/8a/T--Marburg--gg_2dhist2_L34K.png">a</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/8/8a/T--Marburg--gg_2dhist2_L34K.png">a</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/c/c3/T--Marburg--gg_2dhist2_L62K.png">b</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/c/c3/T--Marburg--gg_2dhist2_L62K.png">b</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/0/0a/T--Marburg--gg_2dhist2_R145K.png">c</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/0/0a/T--Marburg--gg_2dhist2_R145K.png">c</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/5/58/T--Marburg--gg_2dhist2_T58K.png">d</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/5/58/T--Marburg--gg_2dhist2_T58K.png">d</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/a/a4/T--Marburg--gg_2dhist2_E65K.png">e</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/a/a4/T--Marburg--gg_2dhist2_E65K.png">e</div> |
| − | <div><img src="https://static.igem.org/mediawiki/2018/8/88/T--Marburg--gg_2dhist2_V147K.png">f</div>
| + | <div><img src="https://static.igem.org/mediawiki/2018/8/88/T--Marburg--gg_2dhist2_V147K.png">f</div> |
| | </div> | | </div> |
| − | <figcaption><b>Figure X:</b> LOREM IPSUM</figcaption>
| + | <figcaption><b>Figure 12:</b> 2D Histogram Lysine-Malate Distance vs Dihedral angle 2. a) L34K enzyme mutant b) R145K enzyme mutant c) V147K enzyme mutant d) L62K enzyme mutant e T58K enzyme mutant f) E65K enzyme mutant</figcaption> |
| | </figure> | | </figure> |
| | <p> | | <p> |
| − | There are multiple interesting things that we can conclude from these graphs.
| + | We can observe high densities for R145K (Figure 11b), which means that the single state it is in is also very stable. |
| − | We can see that R145K shows very high densities, which means that the single state it is in is also very stable.
| + | The very stable dihedral angles indicate an entropically unbeneficial conformation of the side chain. |
| − | Due to the restrained formation entropically this is not very favorable, which further declassifies this variation.
| + | For T58K we observe a stable conformation at a low distance, but also that it is stable at a broad range of distances. |
| − | T58K showed a stable conformation at a low distance, but we can also see that it is stable at a broad range of distances.
| + | L34K has multiple stable angles for dihedral angle 2, whilst having only one for dihedral angle 1 and a small and low distance range. |
| − | L34K has multiple stable angles for dihedral angle 2, whilst having only one for dihedral angle 1 and a small and low distance range.
| + | If we take the 1d as well as the 2d histograms into account, L34K seems to be the most promising of all of the different mutated enzymes. |
| − | If we take the normal as well as the 2d histograms into account, L34K seems the most promising of all of the different variations.
| + | |
| | | | |
| | </p> | | </p> |
| | | | |
| − | <div class="skipTarget" skipname="Wetlab Structural Model"></div>
| + | <div class="skipTarget" skipname="Wetlab Structural Model"></div> |
| | <h3>Wetlab</h3> | | <h3>Wetlab</h3> |
| | | | |
| | | | |
| − | <div class="skipTarget" skipname="Summary Structural Model"></div>
| + | <div class="skipTarget" skipname="Summary Structural Model"></div> |
| | <h3>Summary</h3> | | <h3>Summary</h3> |
| | | | |
| | <p> | | <p> |
| − | We have developed an <i>in silico</i> workflow to design a novel decarboxylase that with minor changes can be adapted for other carbon-carbon bond cleavage enzyme designs, <i>de novo</i> or not.
| + | We have developed an <i>in silico</i> workflow to design a novel decarboxylase that with minor changes can be adapted for other carbon-carbon bond cleavage enzyme designs, <i>de novo</i> or not. |
| − | We predicted that the activation barrier for the reaction is too high for successful catalysis and this prediction was in agreement with our experiments. | + | We predicted that the activation barrier for the reaction is too high for successful catalysis. |
| − | Because of time and resources we were only able to look at single point and double mutations, but the reduction in the activation barrier when using lysine to interact with the carboxy functions showed that there are possibilities to change the electronical structure of the substrate in a meaningful way. | + | Because of time and resources we were only able to look at single point and double mutations, but the reduction in the activation barrier when using lysine to interact with the carboxy functions showed that there are possibilities to change the electronic structure of the substrate. |
| | With the covalent bond between the zinc cation and the substrate we also showed that we can and should dare to think outside the box to find novel ways that could help to create a novel enzyme mechanism. | | With the covalent bond between the zinc cation and the substrate we also showed that we can and should dare to think outside the box to find novel ways that could help to create a novel enzyme mechanism. |
| | With the data obtained through our <dfn data-info="Molecular Dynamics">MD</dfn> Simulations we showed that there are multiple positions in the binding pocket of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> where we can introduce point mutations that interact with the substrate. | | With the data obtained through our <dfn data-info="Molecular Dynamics">MD</dfn> Simulations we showed that there are multiple positions in the binding pocket of <dfn data-info="Acetolactate Decarboxylase">ALD</dfn> where we can introduce point mutations that interact with the substrate. |
| − | <br>
| + | </p> |
| − | <br>
| + | |
| | + | <h3>Outlook</h3> |
| | + | |
| | + | The next steps that we want to perform <i>in silico</i> are that we further explore possibilities of multiple mutations (and therefore multiple sidechains in the binding pocket). |
| | + | We have shown that the interaction of charged side chains, in our case lysine, can lower the activation energy. |
| | + | We think multiple sidechains with the possibility of building salt bridges or hydrogen bonds with malate could alter the electronic structure even further. |
| | + | We have also shown that the nor_1 system (with zinc cation) has only an about ~6kcal/mol smaller activation barrier than the complex (without zinc cation). |
| | + | Even though the zinc cofactor offers the opportunity to stabilize malate in the binding pocket in a very specific conformation it is probably worthwhile to investigate a more <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> style binding pocket without a cofactor. |
| | + | <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> speeds the reaction of the natural substrate up by a factor of 10^17 without a cofactor and is a fascinating enzyme because of that. |
| | + | We strongly believe that there is a possibility to design a binding pocket capable to catalyze this reaction using a <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> style binding pocket. |
| | + | We do not need to engineer an enzyme capable of such a reaction speedup as in <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn>, it is fully sufficient to make an enzyme that is capable to catalyze measurable quantities and improve it using our metabolic engineering workflow. |
| | + | Due to the vast differences between the substrates the binding pocket of <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> is too big for Malate. |
| | + | The binding pocket could only be utilized for malate if we would introduce multiple changes and maybe the backbone of the protein is not suited to design the functioning, yet unknown binding pocket. |
| | + | It is possible to design a binding pocket without cofactor using the method we established and to design an enzyme starting from the binding pocket we could either try to engineer <dfn data-info="Orotidine 5′-monophosphate decarboxylase">ODCase</dfn> or use a programm like <a href="https://www.rosettacommons.org/software |
| | + | "><abbr title="Rosetta"> Rosetta </abbr></a> to screen for other possible candidates with fitting binding pockets. |
| | + | <br> |
| | + | <br> |
| | <i>"I have not failed. I've just found 10.000 ways that won't work." - <b>Thomas A. Edison</b></i> | | <i>"I have not failed. I've just found 10.000 ways that won't work." - <b>Thomas A. Edison</b></i> |
| − | <br>
| + | <br> |
| − | <br>
| + | <br> |
| − | We strongly believe that it is possible to engineer a malate decarboxylase with the method that we developed.
| + | Overall, even though we were not able to design a working enzyme, we are very optimistic about the prospect of it in the future and think that our results can function as a fundament that others can build on. |
| − | Our data also shows that it might be worth it to think out of the box to do so and that there are multiple things that we can and should question about our catalysis idea.
| + | |
| − | In the coming years and decades, enzyme design will become more and more abundant, sophisticated and easy.
| + | |
| − | We would love to make a contribution towards this trend and do hope that we did this with our work.
| + | |
| | + | |
| | | | |
| − | </p>
| |
| | | | |
| | | | |





{kind=link}

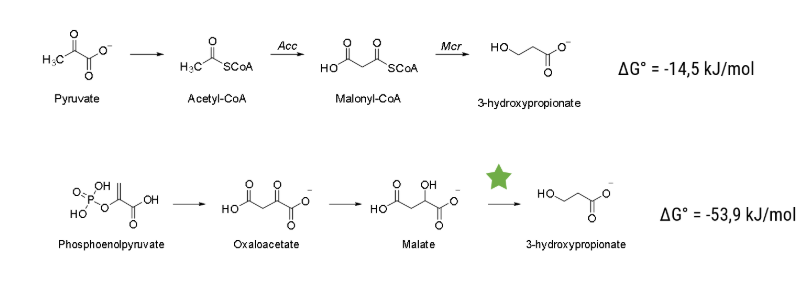

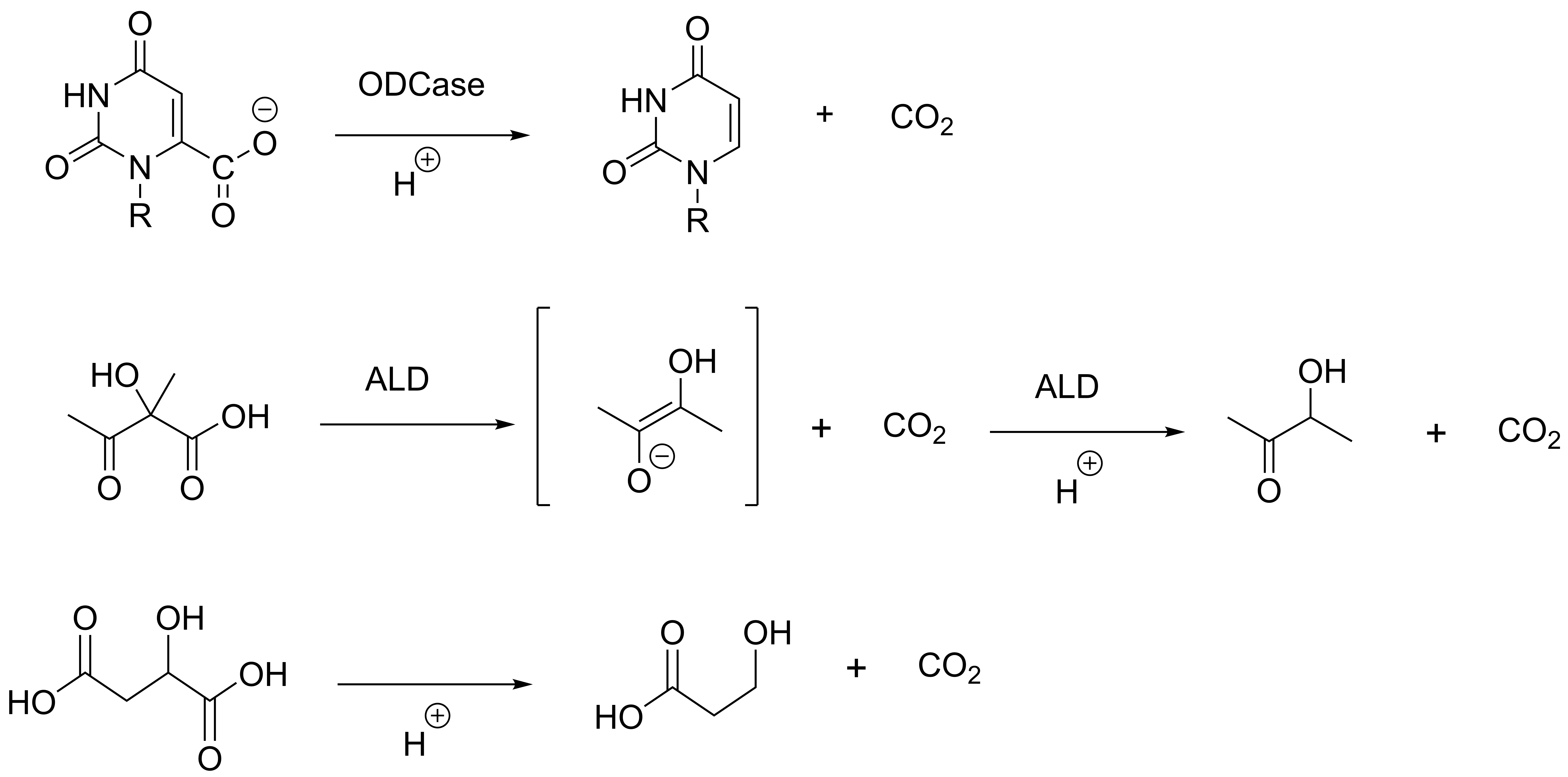
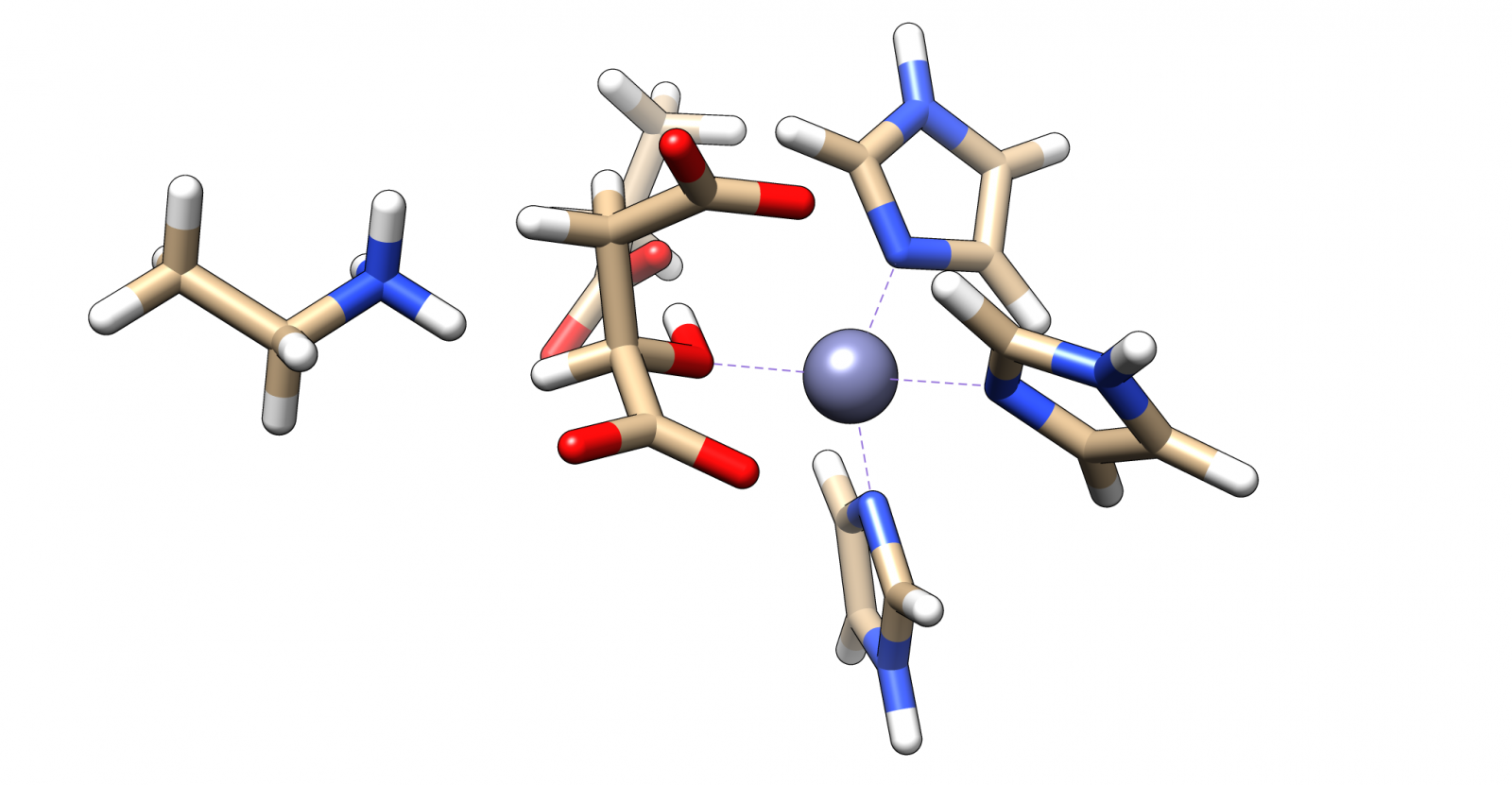 a
a b
b c
c d
d a
a b
b c
c d
d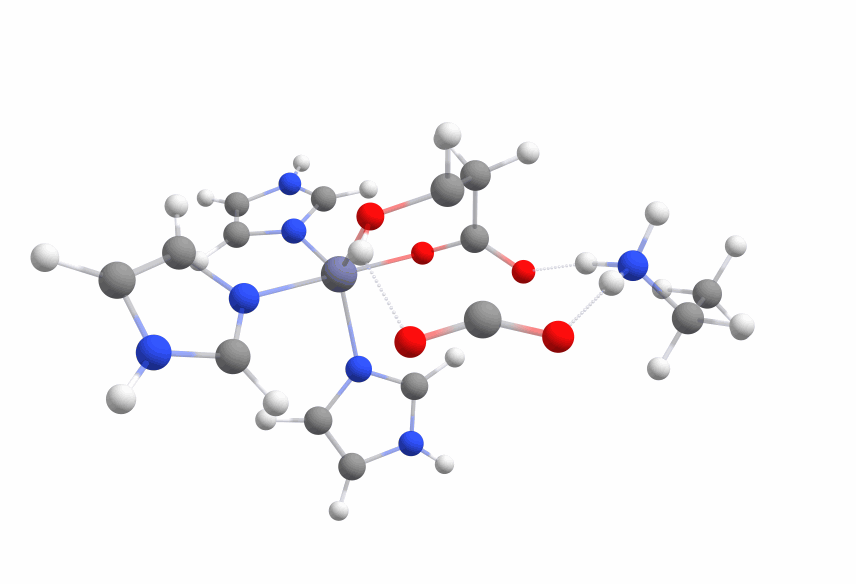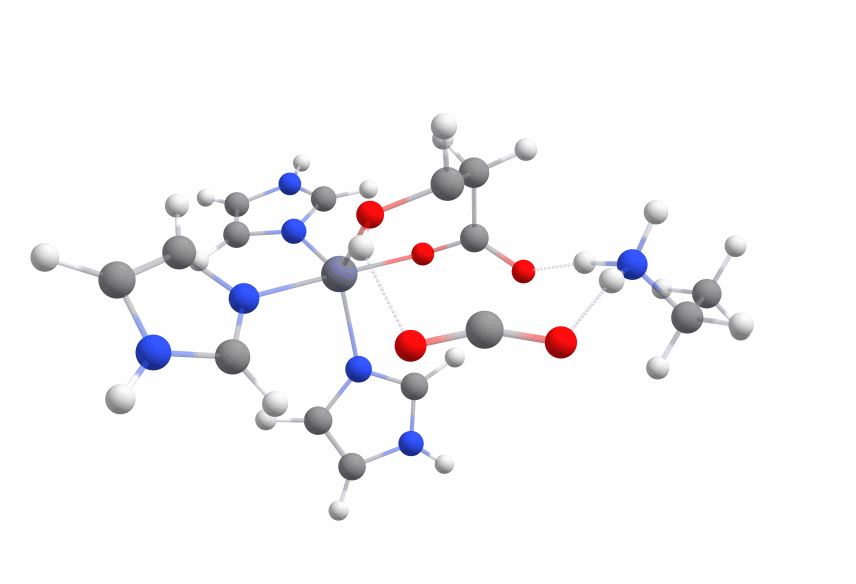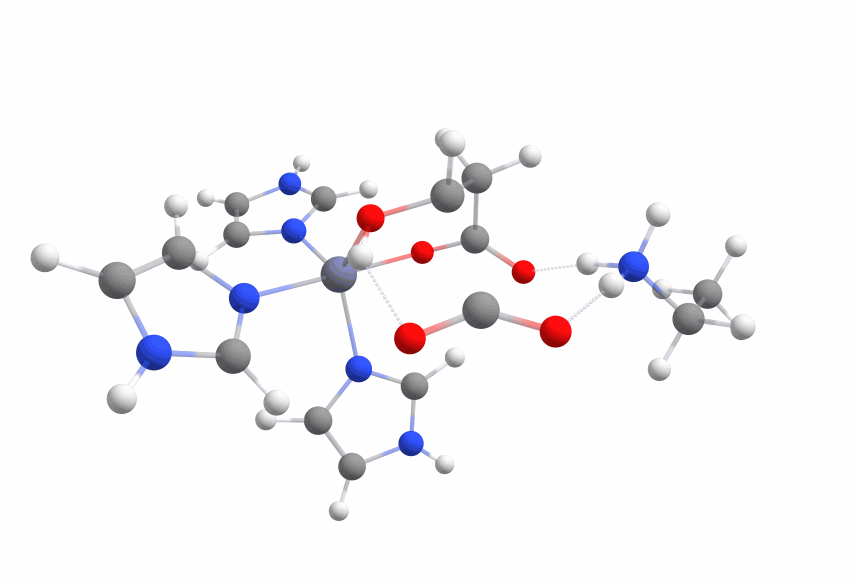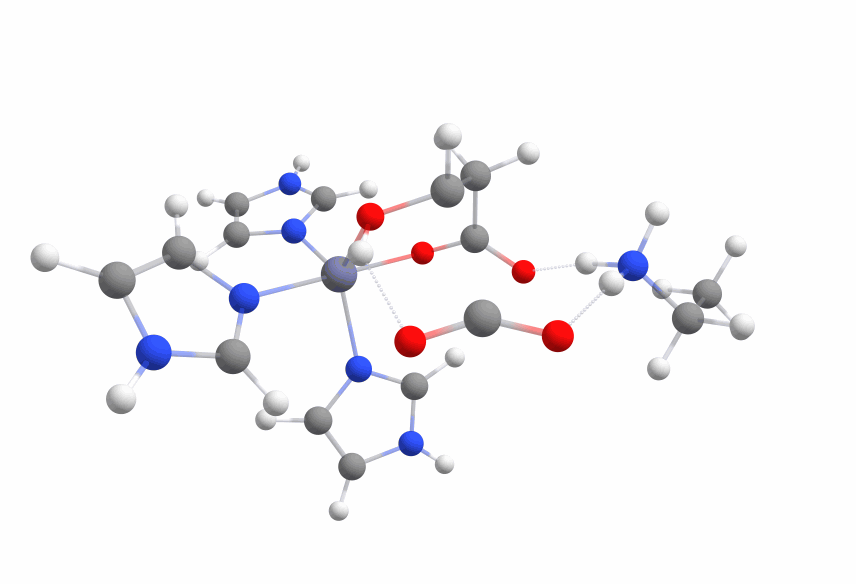
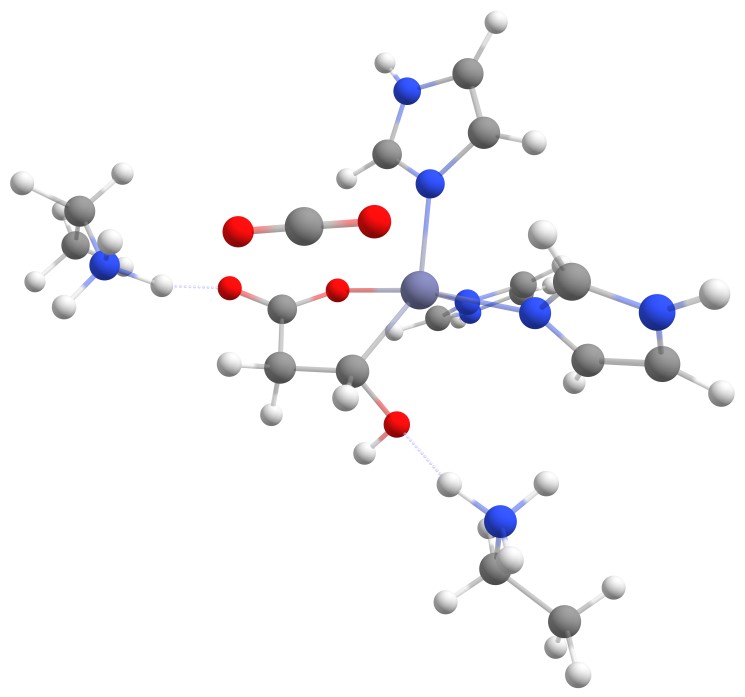

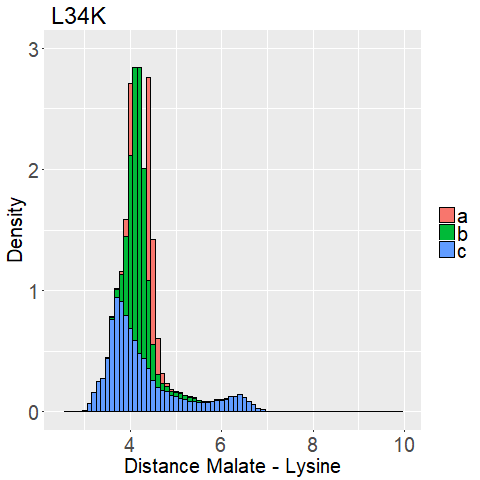 a
a b
b c
c d
d e
e f
f a
a b
b c
c d
d e
e f
f a
a b
b c
c d
d e
e f
f