Team:XJTU-China/Model
Modelling
In order to predict the concentration of different substance in E.coli, we set the kinetic model according to the reaction rate theory and enzymatic reaction kinetics as our first model in our project. And the production and conversion rate model is included to simulate the directed evolution model and the natural evolution model, and then we can get the time we need in our directed evolution method of DTE. The third model we set up is the microfluidics model to predict and simulate the situation in the microfluidics chip, which is our hardware for the gradient concentration of the psicose and antibiotic to let us have a high throughput experiment. The fourth model we set is the market model to predict the future market and the coefficient between different age groups and the tendency to adopt psicose.
- Psicose Synthesis Kinetic Model
- Production Simulink Model
- Market Model
- Microfluidics Model
Psicose Synthesis Kinetic Model
The Establishment of Psicose Synthesis Kinetic Model
In our design, the DTE process is one of the most significant parts in manufacturing psicose. The main process of psicose manufacture is catalyzed by D-psicose 3-epimerase. The models of device A, B, C and D are as follows.
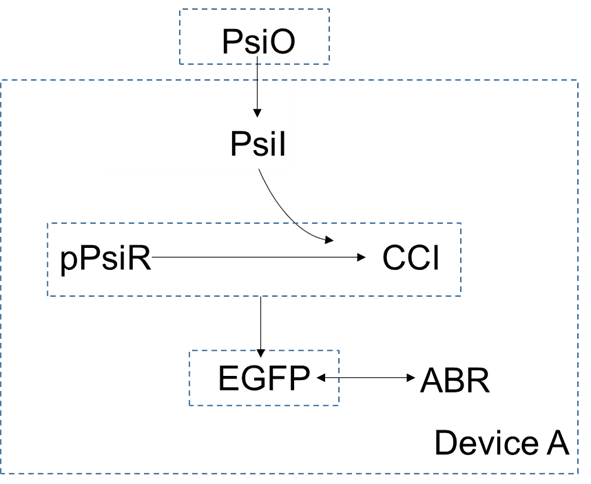
In device A, extracellular concentration of $psicose$ is higher than the intracellular concentration, so it can enter the cells by diffusion. As a small molecular, the $psicose$ inside the cell can be combined with $pPsiR$ to generate $CCI$. $pPsiR$ is a repressor, which can bind to promoters on DNA and block gene expression. After binding with $psicode$,$pPsiR$ falls off from the promoter and the gene starts expressing, And eventually produce the produce $EGFP$.
For device A, the dynamic equations can be listed as follows:
Concentrations of $psicose$ are different inside and outside the cell, so the diffusion rate of $psicose$ is proportional to the concentration difference between inside and outside of the cells.
so the rate at which $psicose$ enters the cell by diffusion is
$$-\gamma_F([PsiO]-[PsiI])$$
Where $\gamma_F$ is the diffusion coefficient.
Considering that the volume of the cells is small enough to be negligible relative to the external solution, so the amount of $psicose$ that diffuses into the cells is very small. We can think of the concentration of the external solution as constant, which means
$$\frac{\text{d}[PsiO]}{ \text{d}t}=0$$
When reducer psicose combines with repressor, the process is
$$\eta_1 PsiI+pPsiR\rightarrow CCI$$
Where $PsiI$ is the intracellular $psicose$, $pPsiR$ is $psicose$ dependent repressor, $CCI$ is $psicose$-repressor complex and $\eta_1$ is coordination number of $psicose$ for $pPsiR$.
The change in concentration of $PsiI$,$pPsiR$ and $CCI$ contains three influencing factors: the binding reaction of $pPsiR$ and $psicose$, and the degradation reaction of themselves and the reverse reaction.
Here we consider the binding reaction as a second order reaction and the degradation reaction and reverse reaction as first order reactions. According to law of mass action, reaction rate is proportional to the product of the reactant concentration, so we have
$$\frac{\text{d}[PsiI]}{\text{d}t}=-\gamma_F([PsiI]-[PsiO])-m_{pPsiR,Psi}[PsiI]^{\eta_1}[pPsiR]+m_{CCI}[CCI]-\delta_{PsiI}[PsiI]$$
And considering $pPsiR$ is constantly expressing, we can get
$$\frac{\text{d}[pPsiR]}{\text{d}t}=\alpha_{pPsiR}-m_{pPsiR,Psi}[PsiI]^{\eta_1}[pPsiR]+m_{CCI}[CCI]-\delta_{pPsiR}[pPsiR]$$
Where $m_{pPsiR,Psi}$ is the coefficient of reaction rate of the binding reaction, $\alpha_{pPsiR}$ is the rate of constant expression of $pPsiR$, $\delta_{pPsiR}$ and $\delta_{PsiI}$ are coefficients of reaction rate of the degradation reaction of $pPsiR$ and $PsicoseI$ respectively.
The concentration of inactivated repressor is
$$\frac{\text{d}[CCI]}{\text{d}t}=m_{pPsiR,Psi}[PsiI]^{\eta_1}[pPsiR]-m_{CCI}[CCI]-\delta_{CCI}[CCI]$$
The change in $EGFP$ concentration depends on the concentration of its repressor, $pPsiR$ which can be described by Hill-equation. Considering $EGFP$ is also degrading, the equation is
$$\frac{\text{d}[EGFP]}{\text{d}t}=H\frac{\beta_{EGFP}K^n}{K^n+[pPsiR]^n} -\delta_{EGFP}[EGFP]$$
Where $n$ is hill coefficient, $K$ is the ligand concentration producing half occupation, $\beta_{EGFP}$ is maximal transcription rate of gene $EGFP$, and $H$ is a constant used to indicate the deviation between the theoretical and actual values.
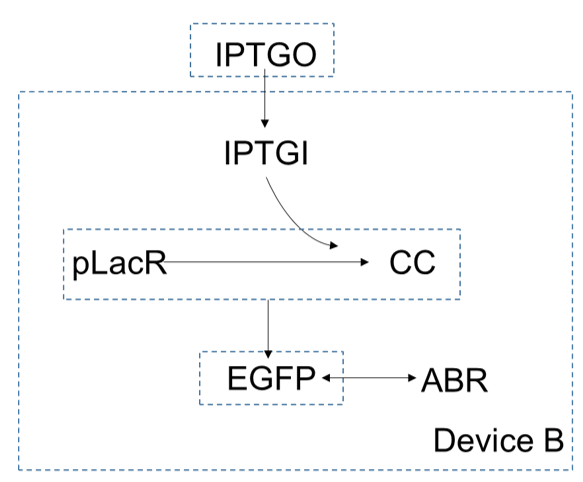
In device B, extracellular concentration of $IPTG$ is higher than which is intracellular, so it can enter the cells by diffusion. As a small molecular, the $IPTG$ inside the cell can be combined with $pLacR$ to generate $CC$. $pLacR$ is a repressor, which can bind to promoters on DNA and block gene expression. After binding with $IPTG$, $pLacR$ falls off from the promoter and the gene starts expressing, and eventually produce the produce $EGFP$. Since the gene of $ABR$ and the gene of $EGFP$ are connected in series, they are expressed together.
Similarly, we can get the function of device B by using the reaction rate equation and the diffusion function. First,
$$\frac{\text{d}[IPTGO]}{\text{d}t}=0$$
According to the reaction between $IPTGI$ and $pLacR$ and the reaction rate equation, we can get
$$\eta_2 IPTGI+pLacR\rightarrow CC$$
Where $\eta_2$ is coordination number of $IPTG$ for $pLacR$.
This reaction is similar with the reaction in device A, so we have
$$\frac{\text{d}[IPTGI]}{\text{d}t}=-\gamma_{IPTG}([IPTGI]-[IPTGO])-m_{IPTG,pLacR}[IPTGI]^{\eta_2}[pLacR]+m_{CC}[CC]-\delta_{IPTG}[IPTGI]$$
Similarly, we can also get
$$\frac{\text{d}[pLacR]}{\text{d}t}=\alpha_{pLacR}-m_{IPTG,pLacR}[IPTGI]^{\eta_2}[pLacR]+m_{CC}[CC]-\delta_{pLacR}[pLacR]$$
$$\frac{\text{d}[CC]}{\text{d}t}=m_{IPTG,pLacR}[IPTGI]^{\eta_2}[pLacR]-m_{CC}[CC]-\delta_{CC}[CC]$$
And $pLac$ is a transcription activator of gene $EGFP$, according to hill equation, the concentration of $EGFP$ is
$$\frac{\text{d}[EGFP]}{\text{d}t}=H\frac{\beta_{EGFP}K^n}{K^n+[pLacR]^n} -\delta_{EGFP} [EGFP] $$
The concentration of $EGFP$ is the same as the concentration of antibiotics resistance due to the transcription and translation of antibiotic resistance gene combined with the gene of $EGFP$.
$$\frac{\text{d}[EGFP]}{\text{d}t}=\frac{\text{d}[ABR]}{\text{d}t}$$
Where $[EGFP]$ is the concentration of $EGFP$ and $[ABR]$ is the concentration of antibiotic resistance protein expression.
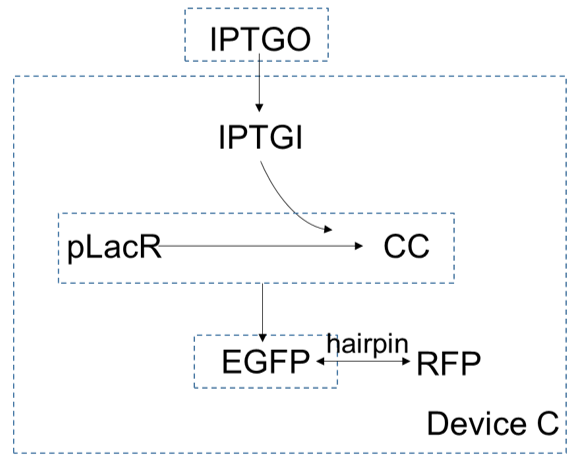
Device C is the same with device B. The only difference is the hairpin between gene of $EGFP$ and gene of $RFP$. Similarly, we can get the function of hairpin and its coefficient.
The first few equations are the same as in device B:
$$\frac{\text{d}[IPTGO]}{\text{d}t}=0$$
$$\frac{\text{d}[IPTGI]}{\text{d}t}=-\gamma_{IPTG}([IPTGI]-[IPTGO])-m_{IPTG,pLacR}[IPTGI][pLacR]+m_{CC}[CC]-\delta_{IPTG}[IPTGI]$$
$$\frac{\text{d}[pLacR]}{\text{d}t}=\alpha_{pLacR}-m_{IPTG,pLacR}[IPTGI][pLacR]+m_{CC}[CC]-\delta_{pLacR}[pLacR]$$
$$\frac{\text{d}[CC]}{\text{d}t}=m_{IPTG,pLacR}[IPTGI][pLacR]-m_{CC}[CC]-\delta_{CC}[CC]$$
$$\frac{\text{d}[EGFP]}{\text{d}t}=H\frac{\beta_{EGFP}K^n}{K^n+[pLacR]^n} -\delta_{EGFP} [EGFP]$$
The presence of the hairpin leads to a decrease in $RFP$ expression efficiency, so we have
$$\frac{\text{d}[EGFP]}{\text{d}t}=k\frac{\text{d}[RFP]}{\text{d}t}$$
Where $[RFP]$ is the concentration of red fluorescence protein, $k$ is the coefficient of hairpin.
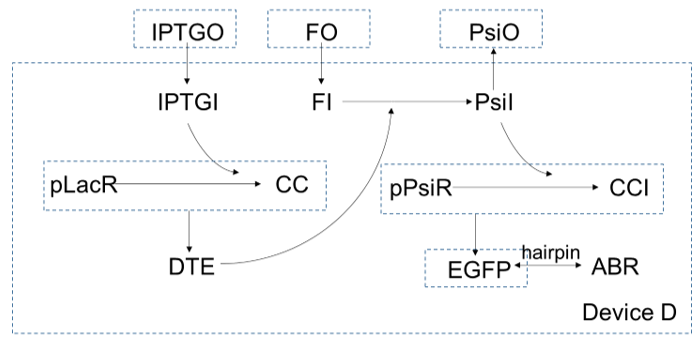
In device D, $IPTG$ gets in cells and bind with $pLacR$, which is a repressor for gene $DTE$. After $IPTG$ binding with $pLacR$, $DTE$ starts to express, and as an enzyme, to catalysis $fructose$ to turn into $psicose$. As more and more $psicose$ are produced, more and more the repressor of gene $EGFP$, $pLacR$ are inactivated, so expression of $EGPF$ increase. At the same time, expression of $ABR$ also increase since the gene of $ABR$ and the gene of $EGFP$ are connected in series by a hairpin.
For device D, $psicose$ and $fuctose$ get in cells by diffusion:
$$\frac{\text{d}[PsiO]}{\text{d}t}=\frac{-\gamma_{F}([PsiO]-[PsiI])}{V_{outside}}=0 $$
$$\frac{\text{d}[FO]}{\text{d}t}=\frac{-\gamma_{F}([FO]-[FI])}{V_{outside}}=0$$
$$\frac{\text{d}[IPTGO]}{\text{d}t}=\frac{-\gamma_{F}([IPTGO]-[IPTGI])}{V_{outside}}=0$$
And device D consists of device A and device C connected by an extra step:
The rest of the equations are the same with which in device A and device C: $$\frac{\text{d}[pLacR]}{\text{d}t}=\alpha_{pLacR}-m_{IPTG,pLacR}[IPTGI][pLacR]+m_{CC}[CC]-\delta_{pLacR}[pLacR]$$ $$\frac{\text{d}[pPsiR]}{\text{d}t}=\alpha_{pPsiR}-m_{pPsiR,PsiI}[pPsiR][PsiI]+m_{CCI}[CCI]-\delta_{pLacR}[pLacR]$$ $$\frac{\text{d}[pPsiR]}{\text{d}t}=\alpha_{pPsiR}-m_{pPsiR,Psi}[PsiI][pPsiR]+m_{CCI}[CCI]-\delta_{pPsiR}[pPsiR]$$ $$\frac{\text{d}[CC]}{\text{d} t}=m_{IPTG,pLacR}[PsiI][pPsiR]-m_{CC}[CC]-\delta_{CC}[CC]$$ $$\frac{\text{d}[CCI]}{\text{d} t}=m_{pSiR}[PsiI][pPsiR]-m_{CCI}[CCI]-\delta_{CCI}[CCI]$$ $$\frac{\text{d}[DTE]}{\text{d}t}=H\frac{\beta_{EGFP}K^n}{K^n+[pLacR]^n}-\delta_{DTE}[DTE]$$ $$\frac{\text{d}[EGFP]}{\text{d}t}=H\frac{\beta_{EGFP}K^n}{K^n+[pPsiR]^n}-\delta_{EGFP}[EGFP]$$ $$\frac{\text{d}[ABR]}{\text{d}t}=k\frac{\text{d}[EGFP]}{\text{d}t}$$ The results are shown in the Results Tab as follows.
Production and Conversion Rate Simulink Model
The Establishment of Production and Conversion Rate Simulink Model
To understand how we get psicose in a deep view, replication, transcription and translation are involved to describe the synthesis of psicose and thus come to our production on a large scale. As we all know, the enzyme will degrade due to the mutation of coding sequence during DNA replication and the transcription error.
Although the rate mutation and transcription error are one in 100 million and one in 10 thousand, the yield of psicose may not decrease because even errors happen in the process of replication and transcription, the type and order of amino acid may not reverse as well as the function of our enzyme which lead to the same catalytic rate of the original one.
However, the butterfly effect tells us that we cannot ignore the small change in our system, even it is little enough for us to ignore. Considering the mutation rate and transcription error, the production of psicose is closely related to the cycling times, error rate and original production. The production of psicose is a function with variables of cycling times, error rate and original production.
$$Prd(t)=Prd(t,Times,errRate,oriPrd)$$
Where Times is the cycling times of psicose, errRate is the rate of replication and transcription error, oriPrd is the original production of our system.
The errRate is influenced by the replication error rate and the transcription error rate. Meantime, the more replication and transcription errors we get, the lower or higher production we will get. The error rate function is
$$errRate(t)=repRat(t)\cdot trsRat(t)$$
Where the replication error rate depends on the length of D-psicose 3-epimerase coding sequence, the multiplication of single error rate define the replication and transcription rate as follows
$$repRat(t)=srepRat^{len}(t)$$
The length of mRNA is one third of the cDNA length, here we get
$$trsRat(t)=\sqrt[3]{strsRat^{len}(t)}$$
Although errors can result in the decrease of production, the increase of production is also promoting on account of the uncertainty of variation. For a natural evolution system, we consider that the probability of production decrease and increase is equal, decided by the error rate per cycling period. Therefore, the average production growth rate is distributed normally with the mean of 0 and the variance of error rate. The production growth rate is shown as follows
$$grsRate \sim N(0,errRate)$$
If we set X as production growth rate,$\sigma^2$ as error rate, then we will get
$X \sim N(0,\sigma^2)$
And the probability density function is
$$f(x\mid 0,\sigma^2)=\frac{1}{\sqrt{2\pi\sigma^2}}e^{-\frac{x^2}{2\sigma^2}}$$
The probability of production growth rate is
$$P(x_1 < grsRat < x_2)=F(x_2)-F(x_1)=\int_{x_1}^{x_2}f(x)dx$$
The original production is also vital when we simulate the production of our system by reason that the larger our system is, the more psicose we will get in our manufacturing and it makes no sense to compare a certain system with another in a totally different scale. So the standardization and the estimation of original production for psicose is necessary.
There are many variables which have impacts on the original production, such as the quantity of E.coli, the growth rate of E.coli, the conversion rate of psicose and the total amount of substrate which determines the scale of the production system. The original production rate is as follows
$$\frac{\text{d}oriPrd}{\text{d}t}=N\alpha C_{sub}$$
Where $oriPrd$ is the original production of the psicose production system, $N$ is the number of E.coli in the system, $\alpha$ is the conversion rate for D-psicose 3-epimerase, $C_{sub}$ is the concentration of substrate, which represents the concentration of fructose in our production system for psicose.
As for the growth of E.coli, we use Logistic equation to describe the process of E.coli proliferating.
$$\frac{\text(d)N}{\text{d}t}=r_1N(1-\frac{N}{r_2})$$
Microfludics Model
The Establishment of Microfludics Model
In our Lab tour, we find that it is extremely complicated to use pipette to prepare and transfer solutions, especially gradient concentration solutions. High throughput methods to get the gradient concentration of solutions are well needed. In this case, we made a hardware by using the principle of microfluidics and then we simulate it whether it can give us different concentration by using the microfluidics device before the microfluidics chip is finally made.
To get the downstream concentration of high concentration and low concentration, particle collision is used to demonstrate the downstream concentration.

When the fluid flows into the vertical channel of the microfluidics chip, the number of the solute is equal and both are half of the original quantity. In this case, the concentration of the both sides or both directions are the same with the half original concentration.
$$m_3=m_4=\frac{1}{2}m_1\qquad m_5=m_6=\frac{1}{2}m_2$$
$$n_3=n_4=\frac{1}{2}n_1\qquad n_5=n_6=\frac{1}{2}n_2$$
$$c_3=c_4=c_1\qquad c_5=c_6=c_2$$
When the liquid flows to the corner of the channels, the momentum is considered as a constant due to the orthogonal relation between the vertical channel and the horizontal channel. According to the momentum conservation theorem, we can get
$$m_4\overrightarrow{v_1}+ m_5\overrightarrow{v_2}=(m_4^{'}+m_5^{'})\overrightarrow{v_3}+\int{f(n,l)dt}$$
Regard the process of two solutions converging together as particles colliding, most particles changes their direction of motion, and flow vertically down. While some particles still remain inside the tube. Take these as the loss at time $t$.
Assume the probability function of collision loss is $p(n)$, for the loss is in proportion with the amount of particles, the convergence concentration $c_7$
$$c_7=(1-p(n_2))+c_2(1-p(n_1))$$
Hence the local concentration can be determined by the former concentrations. Assume the particle amounts as $n_7$
Take $ m=knc$ into equation 1, we can get
$$m_4\overrightarrow{v_1}+ m_5\overrightarrow{v_2}=kn_7c_7\overrightarrow{v_3}+\int{f(n{4,5},ll)dt}$$
For the channel at the first column at each row, $$ c(i, 1)=c(1,1)$$ For channels the last column at each row, $$ c(i, i+1) = c(1,2) $$ For the concentration at a certain exit $$ c(i, j)= c(i-1,j-1)\times (1-p(n(i-1,j)) + c(i-1,j) \times (1-p(n(i-1,j-1)) $$ Then by the iterative approach, we can calculate the value of $n$, which is the constraint for the corners and crosses of the channels $$n(i,j)=\frac{m(i-1,j-1)\times \vec{v}+m(i-1,j)\times \vec{v}-\int f(n,l)dt}{kc(i,j)\vec{v}}$$ To solve our model ,the Navier-Stokes Equations are adopted with the assumption of momentum conservation and mass conservation.

Assumptions:
1. Newton Fluids: incompressible, its density and viscosity unaffected by concentration change
2. The channel is smooth and the phase change tension can be ignored
3. For the liquid near the channel walls, the velocity in y direction and z direction are 0, and x direction are not zero
4. Due to the microfluidics phase is mainly by the features of the solution, the grativity is ignored and the internal force is ignored.
Then we can get a set of equations:
$$\rho\frac{\partial v}{\partial t}+\rho v \triangledown v=-\triangledown P+\mu \triangledown ^2 v$$ Where, $\rho$ is the density of the solution, $v$ is the velocity, $P$ is the pressure intensitys of the system,$\mu$ is the viscosity of the liquid,$\triangledown$ is Hamiltonian operator: $$\triangledown =\frac{\partial}{\partial x} i+\frac{\partial}{\partial y} j \frac{\partial}{\partial z} k $$ We can get the velocity vector based on knowing the pressure intensity of a certain point in the channel. We apply the velocity vector into the C-D equation, then we can get the concentration $c$ of a certain point: $$\frac{\partial v}{\partial t}+Dv\cdot \triangledown c=D\triangledown^2 c$$ Where $D$ is the diffusion coefficient of the liquid and $c$ is the concentration of the solution. Taking $m,n,c$ we have mentioned, we can get the concentration distribution as follows:

Potential Market Model
The Establishment of Potential Market Model
In our market model, we’d like to analyze the relationship between the choice of psicose and the market scale. Then, the market od psicose is predicted.
Results and Discussion
Basing our model, we can calculate and simulate our system as follows:
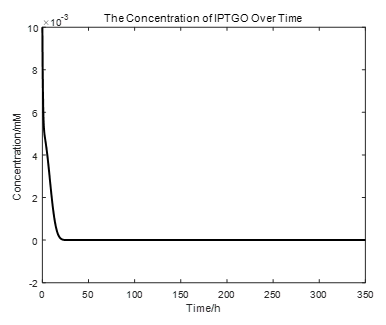 |
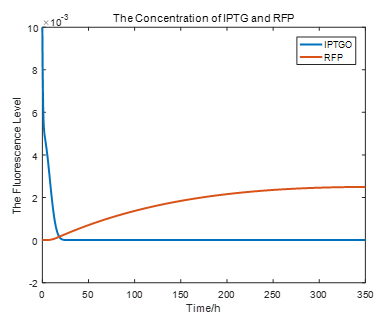 |
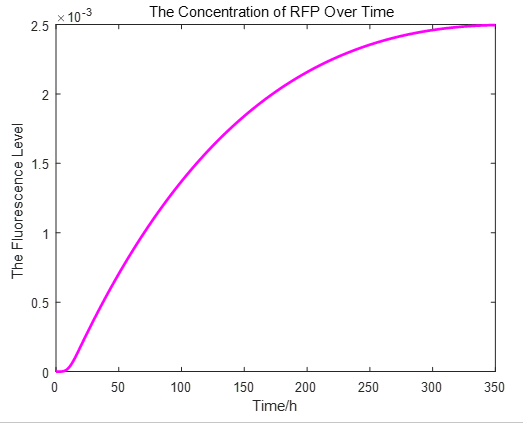
From the picture below, it is evident that with time going by, the concentration of IPTG outside the cell will decrease due to the diffusion process, while the RFP expression level is increasing stably.
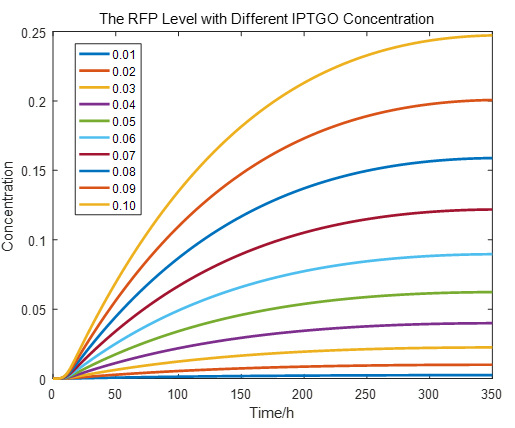 |
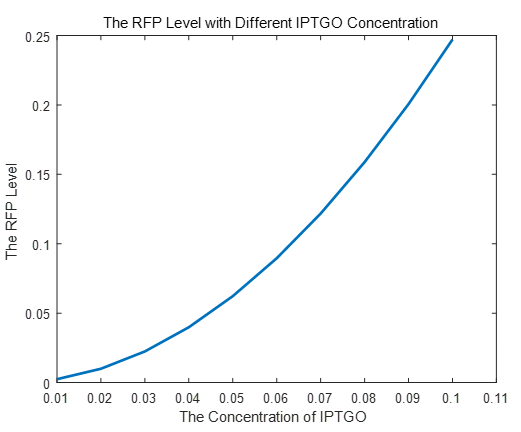 |
If we do more experiments shown below, we can find that the expression level of RFP is increasing with the increase of the concentration of IPTG, which proves that we can get a linear range of IPTG to get the RFP expression level. According to device A, B, C, D, this conclusion can also be made, which proves the validation of our kinetic model
 |
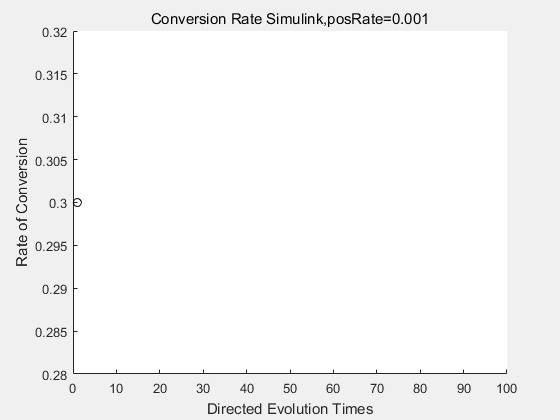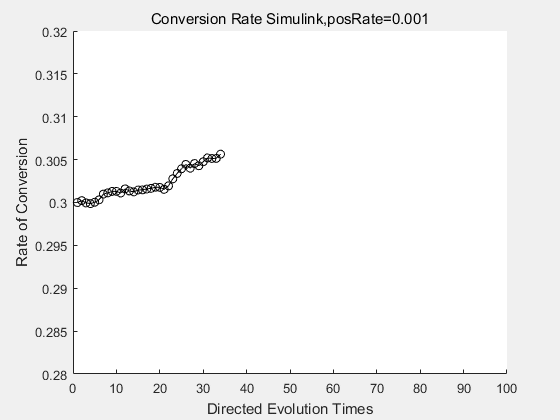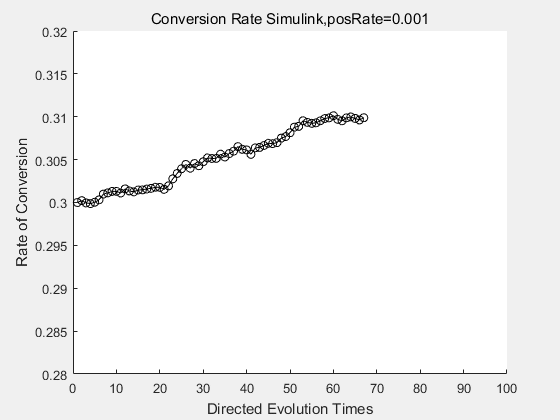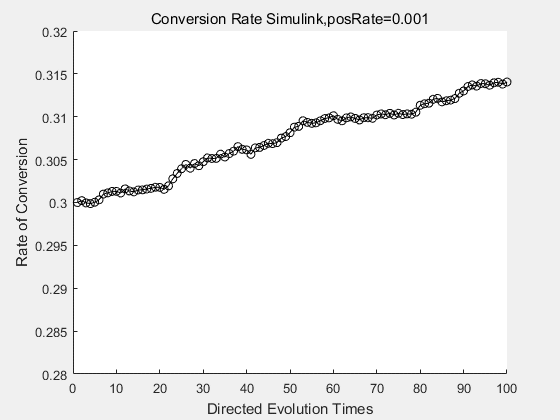 |
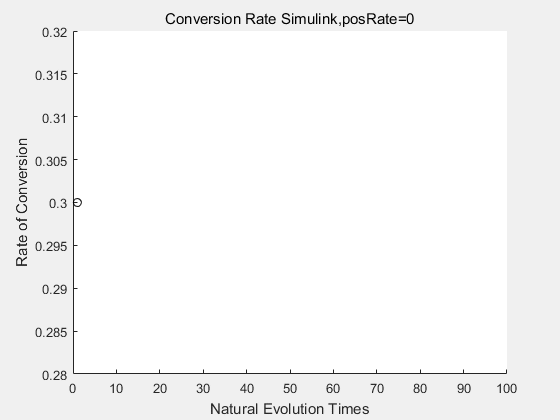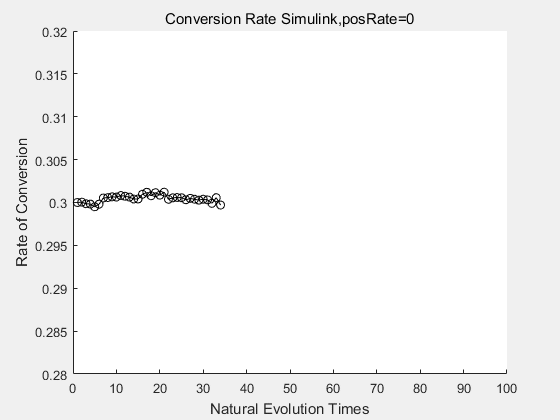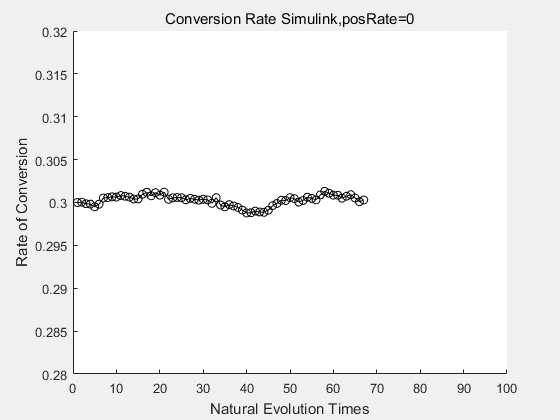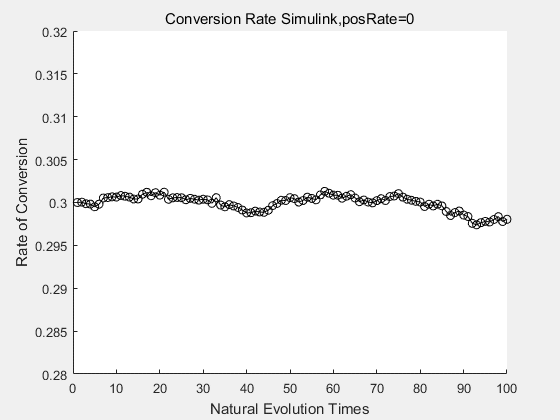
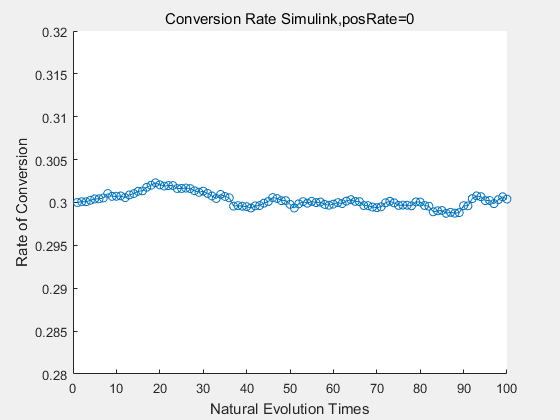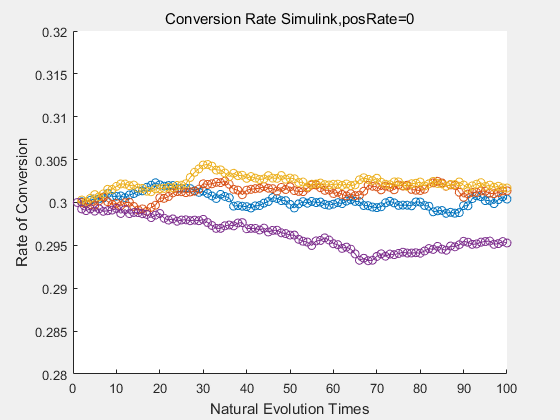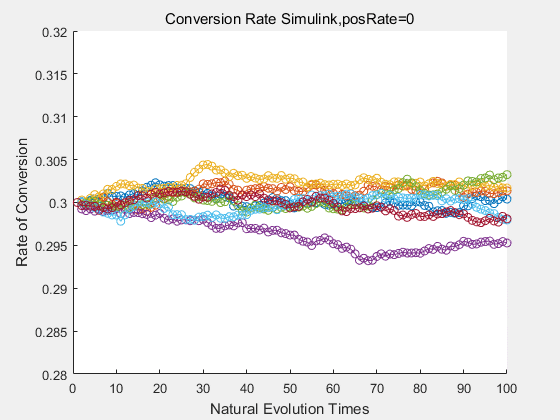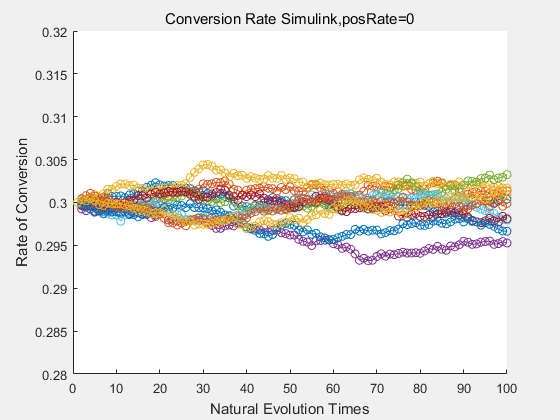
With the directed evolution method to produce psicose, what matters is the conversion rate and the scale of the production system. As the results shown above, if we cultivate our system for many times, the conversion rate of DTE is increasing stably with the directed evolution method.
 |
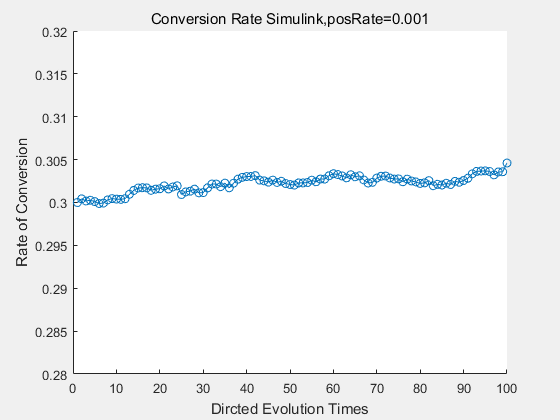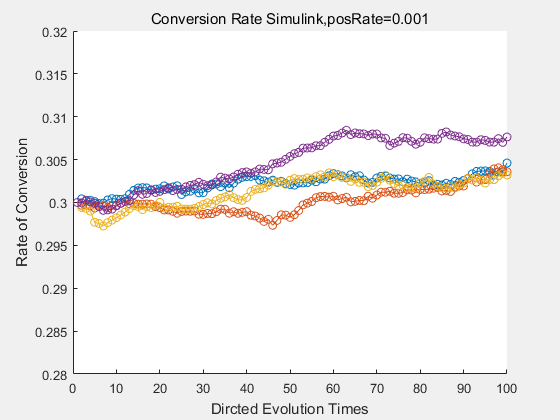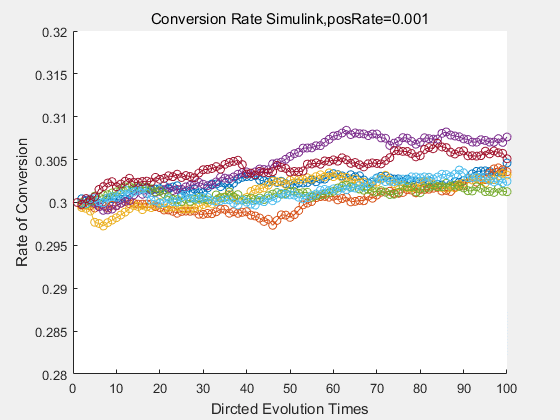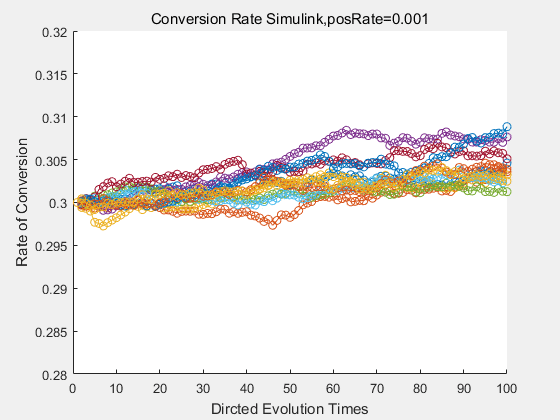 |
If we do diretecd evolution for more times, we will get the same result and the increase of conversion rate with directed evolution is stable, while the conversion rate of DTE in natural evolution system fluctuate in a certain range.
 |
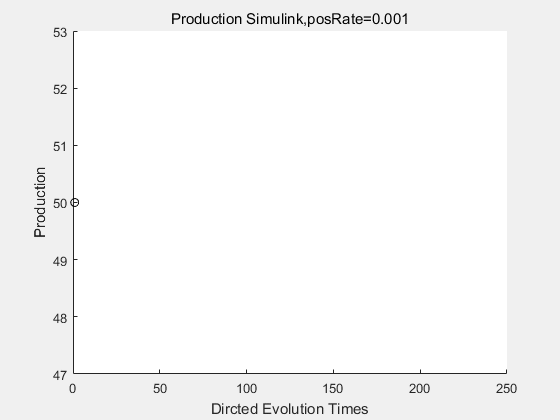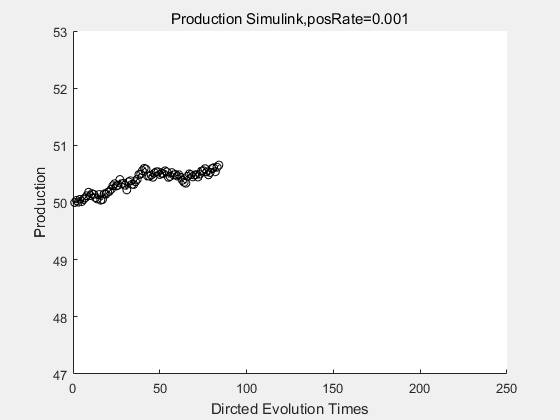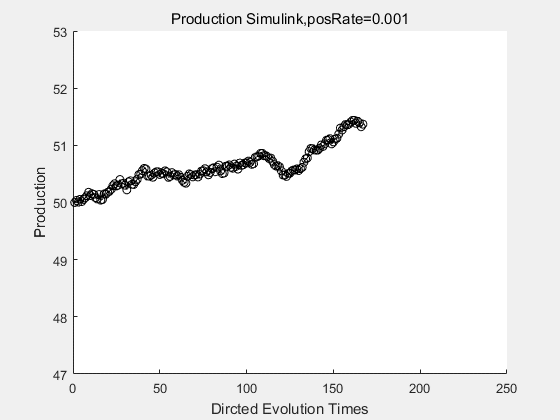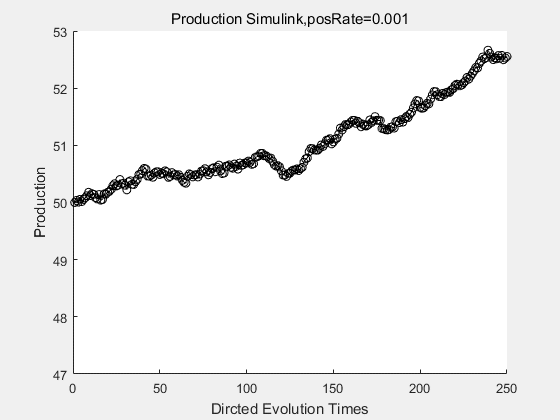 |
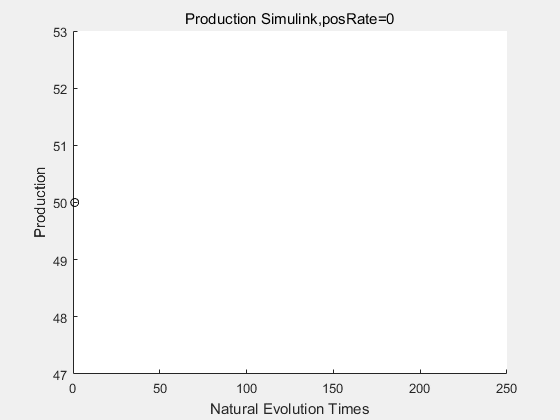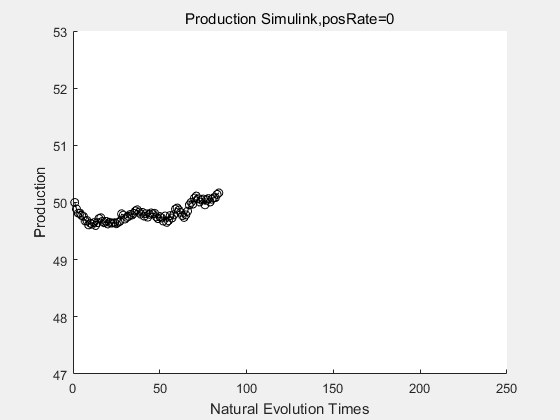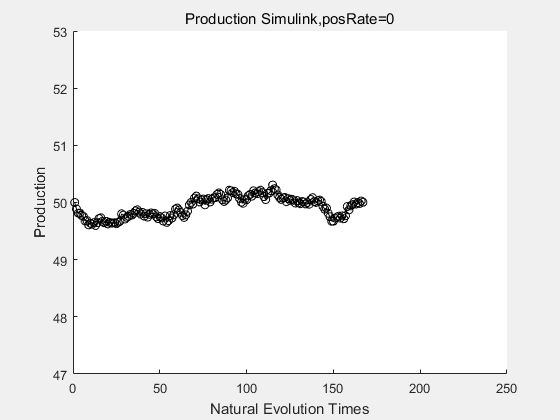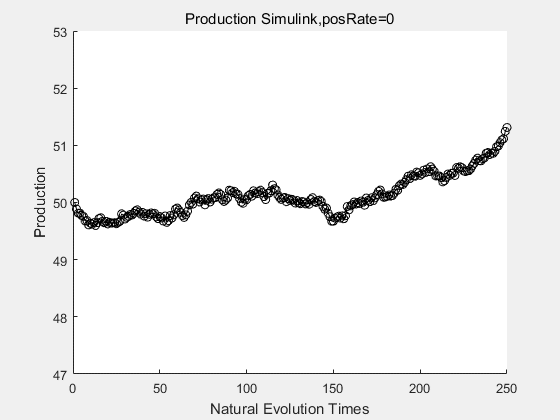
If we have a system with a stable psicose production of 50,what happens in directed evolution system is the slight increase in the production with the times of producting increasing. Whereas, the production of natural evolution system is still in the original production and vibrate in a small range.
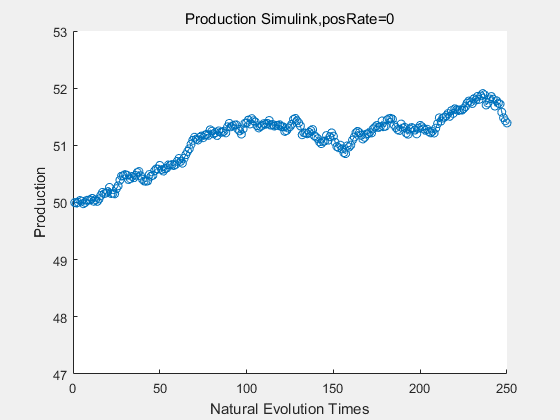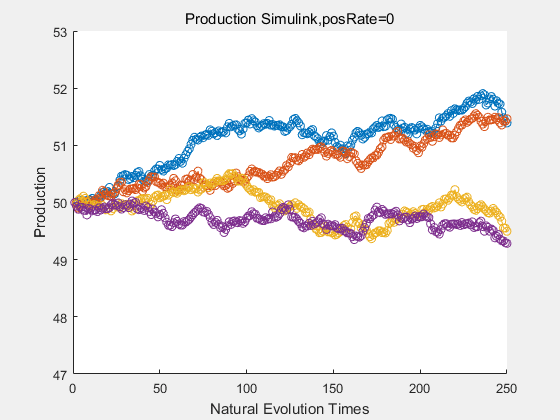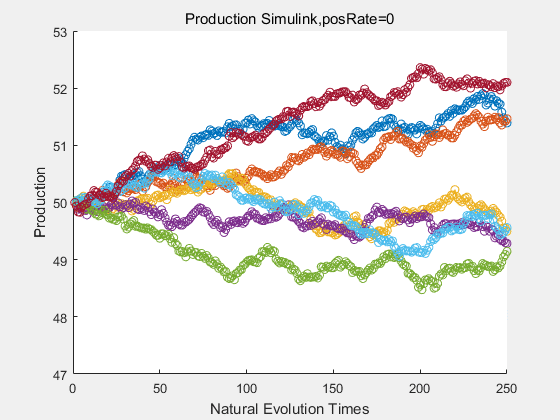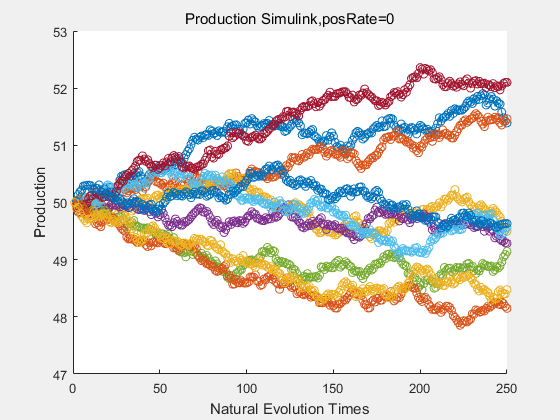 |
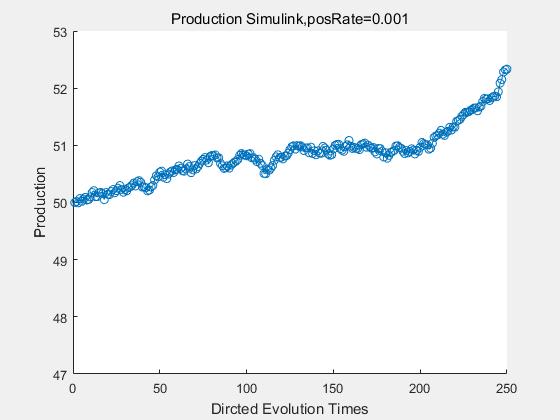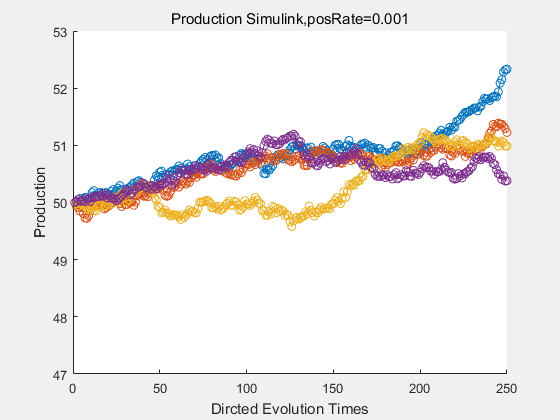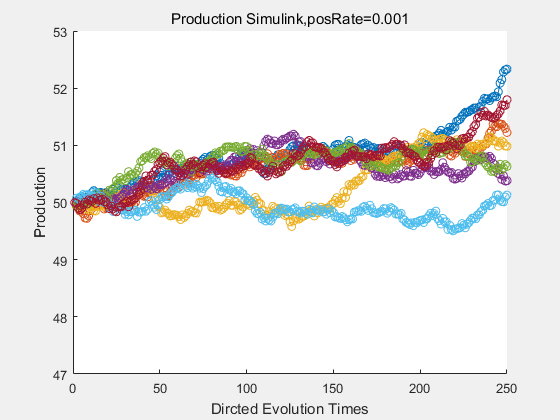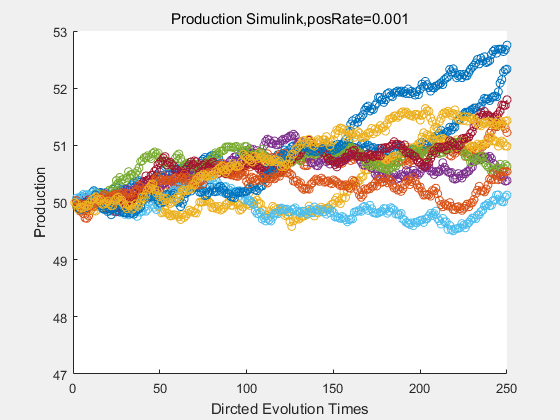 |
With more simulations ,we can find that although the production is not always increasing all the time, the main stream of the directed evolution is to have a good production of psicose. The production of natural system will increase or decrease due to the vibration of the conversion rate, and if we select the positive strain each evolution process,, we will get the same production results which is the factory method for strain selection.
Reference
Schmidt F R. Optimization and scale up of industrial fermentation processes.[J]. Appl Microbiol Biotechnol, 2005, 68(4):425-435.
Lin P Y, Whang L M, Wu Y R, et al. Biological hydrogen production of the genus Clostridium: Metabolic study and mathematical model simulation[J]. International Journal of Hydrogen Energy, 2007, 32(12):1728-1735.
Whang L M, Hsiao C J, Cheng S S. A dual-substrate steady-state model for biological hydrogen production in an anaerobic hydrogen fermentation process[J]. Biotechnology & Bioengineering, 2010, 95(3):492-500.
Rousu J, Elomaa T, Aarts R. Predicting the speed of beer fermentation in laboratory and industrial scale[J]. 1999, 1607.