Team:Macquarie Australia/Results
To summarise and validate the successful construction of the parts comprising the Chlorophyll Biosynthesis Plasmid, all composite parts underwent single (EcoRI) and double (EcoRI with PstI) digests followed by agarose gel (0.5%) electrophoresis. The results from the digests showing the successful assembly of the Chlorophyll Biosynthesis Plasmid are shown in Figure 1 (below).
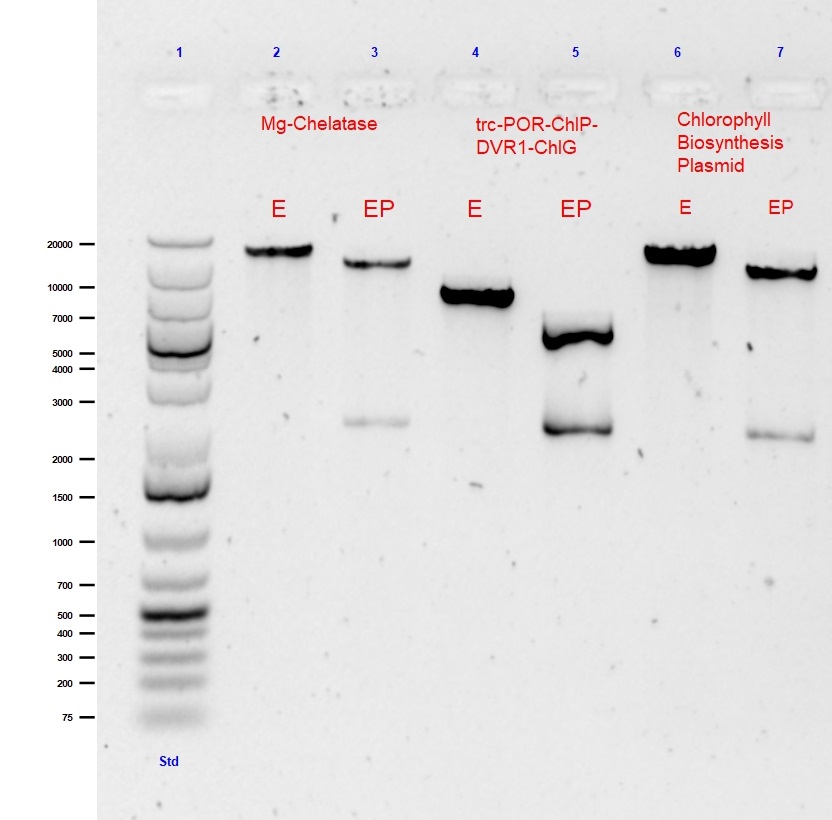
Fig. 1 Agarose gel (0.5%) electrophoresis with GelRed of single (EcoR1) and double (EcoR1 with PstI) digests of submitted composite parts: Mg-chelatase plasmid (9513bp), trc-POR-ChlP-DVR1-ChlG (4604bp) and Chlorophyll A Biosynthesis pathway (18352bp).
This large 9.3kB biobrick of 5 genes was created to begin the 1st step of the Chlorophyll biosynthesis pathway away from Heme biosynthesis utilising the cells naturally produced PPIX to Mg-PPIX. Standard assembly of trc-ChlI1-ChlI2-ChlD and trc-ChlH-GUN4 was performed in a CAM biobrick backbone. The single and double digest of this Mg-chelatase plasmid is shown in lanes 2 and 3 of Figure 1, respectively.
The second large 4.7kB biobrick construct called “PPDG” was created as the final part of our Chlorophyll A biosynthesis. This composite part was created to convert photoxidative PChlide to Chlorophyll A. This was important as it allowed for stability of our light triggered vesicle formation system. This part was formed using Standard assembly of trc-POR-ChlP and DVR1-ChlG into a CAM backbone.The single and double digest of this trc-POR-ChlP-DVR1-ChlG plasmid is shown in lanes 4 and 5 of Figure 1, respectively.
The final construct is the assembly of the whole “CBP Biobrick”. This is 18.3kB composite part was pieced together using all 14 genes of our Chlorophyll biosynthesis pathway into the CAM backbone. This part was formed using Standard assembly of previously assembled Mg-chelatase plasmid and lac-CTH1-ycf54-ChlM-trc-FNR-fdx-trc-POR-ChlP-DVR1-ChlG.The single and double digest of our pièce de résistance CBP plasmid is shown in lanes 6 and 7 of Figure 1, respectively.
The POR gene with the trc promoter showed a high level of inducible expression by IPTG within the KAN backbone (Figure 2). The POR protein is seen clearly at the expected size of 40 kDa. This result validates the expression of POR under the new trc promoter.

Fig. 2 SDS-PAGE gel of trc-POR overexpression with IPTG. POR was identified at 40kDa in both soluble lysate (Lane 3) and insoluble pellet (Lane 4).
Protochlorophyllide (Pchlide) was extracted from barley (Figure 3) and used as a substrate for to assess the function of the expressed POR. The barley extracted Pchlide was added to the lysate of the IPTG induced- E.coli containing the trc-POR construct.

Fig. 3 Protochlorophyllide extracted from barley fluorescing under a UV torch.
The mixture was incubated for varying times (0 to 30 mins) then placed in a spectrophotometer. As shown in Figure 4, an absorbance at 627-630 nm shows the presence of protochlorophyllide. An absorbance peak at 665-670 nm would be expected for POR activity but this could not be seen.We believe a lack of a POR peak at 665-670nm may be due to the presence of other cell proteins in the crude cell lysate that interfere with our assay. Literature on this assay has stated that the assay requires purified POR protein (Townley et al, FEBS Letters 422: 19-22 1988). We continue to work on the purification of our expressed protein using chromatography methods.

Fig. 4 UV vis spectrum of our purified protochlorophyllide from barley. Protochlorophyllide is observated at a peak of 437nm and a maxima of 1.8.
Using a fluorescence spectroscopy assay we checked for production of Mg-PPIX using our induced Mg-chelatase plasmid. The sample was excited with an excitation wavelength of 420nm. Purified proteins of Mg-chelatase (provided by our advisor Professor Robert Willows) was used as a positive control.. A peak appearing at 595nm shows successful production of Mg-PPIX, as demonstrated by our positive control (Figure 5). We assayed our induced Mg-chelatase plasmid by adding purified individual protein subunits of Mg-chelatase to see if the expression of any individual gene was limiting. No added individual protein subunit demonstrated an enhancement on the expression of our Mg-PPIX using our Mg-chelatase plasmid. We proceeded by adding ChlI1,ChlI2 and ChlD to our Mg-chelatase plasmid lysate. This was performed to see if we could characterise the expression of ChlH. This yielded similar results. Our findings are also supported by the qPCR expression results provided by our collaboration with NTU iGEM team that showed an almost complete lack of expression of our genes before and after induction compared to the chloramphenicol control(Results shown in our collaboration page). We are continuing to investigate the reasons for this unexpected lack of expression.

Fig. 5 Fluorescence spectroscopy assay of Mg-chelatase verified proteins (Positive control). Excitation at 420nm was used to view Mg-PPIX formation. Mg-PPIX was seen at a wavelength of 595nm.
We set out to model the production of chlorophyll from aminolevulinic acid based on the kinetic parameters of the enzymes in the system and the realistic ranges of concentrations of those enzymes that would be achievable in an E. coli cell. We looked at what the concentrations of enzymes could be based on the volume of an E. coli cell and what the actual amounts of protein could exist in an E. coli cell based on published literature values.
The model generated was successfully able to model the production of 12 different constituents based of the enzyme kinetics parameters, indicating that our program is robust and capable of predicting complex pathways with many enzymes of varying kinetic parameters. As our program condenses our enzyme parameters, such as Kcat and Km values, into a series of single-line arrays, our script can be easily re-programmed to model other metabolic pathways, making it versatile and flexible. In addition, individual enzyme concentrations can be altered through prompts before running the model, allowing for fast re-programming to quickly generate a set of models, and allow for efficient testing.
Of all enzymes tested, the enzyme that produced the greatest increase in chlorophyll production, when increased, was Magnesium chelatase (Figure 6), which was increased tenfold to 1000nM. By changing only one enzyme, the sustained accumulation of PPIX and all proceeding intermediates was eliminated, producing an extremely efficient biosynthesis pathway that converts almost 100% of the input ALA into chlorophyll within 10 days.

Fig 6. Model with 1000nM concentration of Magnesium chelitase.




