Team:TJU China/Description
<!DOCTYPE >







CRISPR-Cas System
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat) system, which forms the adaptive immune system in bacteria,
has been modified for genome engineering. In this system, an endonuclease (Cas protein) is guided by a short sequence
of RNA (gRNA) to bind to the gene of interest and cut it. Modifications of various Cas enzymes have extended CRISPR
to activate/repress target genes selectively. Moreover, one can change the genomic target of the Cas protein by simply
changing the target sequence present in the gRNA. Due to its comparative simplicity and adaptability, CRISPR has
rapidly become the most popular genome engineering approach.
There are different kinds of applications of CRISPR. So far, scientists have achieved gene editing in cells, detection
of diseases etc. using the CRISPR system. We respect the achievements of them, and explore the room of improvement
at the same time. After reading lots of literatures and brainstorms, we focus on four parts of CRISPR system. In
our project, we improve the sensitivity of circuit for detecting heavy metal ions making use of the ability of enhancing
transcription of dCas9. According to Cas12a’s non-specific cleavage properties after specific cleavage, we use it
to not only achieve but also optimize cell-free cancer detection. In addition, to improve the efficiency of in vitro
delivery of CRISPR system, we use our special non-viral vector to deliver the RNPs (complex of sgRNA and Cas9 protein)
into cells. Finally, we get to think about turning to a new area for CRISPR system-- curation of mitochondrial diseases.
In a word, we have tried our best to improve the sensibility and gene-editing efficiency of the system itself as
well as to utilize the system to enhance other biological approaches.
High-throughput disease detection based on CRISPR-Cas12a
Background
As is known to all, the innovation of crisper technique is conducting gene editing by protein-RNA complex. CRISPR-Cas12a
(Cpf1) proteins are RNA-guided enzymes that bind and cut DNA as components of bacterial adaptive immune systems.
Nowadays,Jennifer A. Doudna’s leading team[1] insists that unleashing the ssDNase activity of Cas12 proteins offers
a new strategy to improve the speed, sensitivity and specificity of molecular diagnostic applications. Our program
just uses sequence-specific DNA cleavage of Cas12a to recognize sequence-specific DNA in vitro or in cfDNA and then
non-specifically recognize and cleavage fluorescence probes, which achieve cancer detection in vitro by reflecting
indirectly with fluorescence values.
Trouble shooting
Due to the diversity of mutation sites in human genes, not all mutation sites have suitable PAM (TTTN). However, Cas12a
protein needs PAM to perform recognition function, so the detection of mutation sites is not completely detectable.
Moreover, most mutation sites are very close to each other, and one crRNA can only detect one mutation. Considering
different mutation sites in human genes correspond to different diseases, the detection is expensive. It is necessary
to achieve one-time multi-locus mutation detection as efficiently as possible.
What’s our solution?
In order to solve the problem mentioned above, we, inspired by Wang Jin[2], detected the designed point mutations on
the 150bp PCR fragments, and since the primers includes TTTN (PAM sequence recognized by Cas12a), the target sequences
can be cleaved by Cas12a, therefore we realized no difference detection. According to the rule of arrangement and
combination in mathematics, all the crRNA are designed to avoid errors in the detection results. In view of the efficiency
and cost of detection, we imagine that if the Cas12a protein can be spread on the chip in order, we can then use
the least amount of protein to achieve the most efficient detection, which is called high-throughput detection. So
we tried to dry the hydrophobic protein ( an amphiphilic protein which has a hydrophobic patch as well as hydrophilic
surface ) on the chip firstly, and then the fluorescence probe was cut by the cas12a protein and crRNA complex. Surprisingly,
We found that we detected higher fluorescence values under the condition of a smaller amount of protein.

Targeted delivery of sgRNA/Cas9 complex
Background
CRISPR system has been widely used as a means of gene editing nowadays. And compared to the transfection of plasmids
that encoding Cas9 protein and sgRNA(single guide RNA) or to the delivery of the Cas9 mRNA and sgRNA into target
cells, direct delivery of the Cas9 protein complexed with sgRNA is the most widely studied strategy in recent years.
The purified Cas9 protein is positively charged and can efficiently form a complex with sgRNA, which is called Cas9/sgRNA
ribonucleoprotein complexes (RNPs). Direct delivery of RNPs has numerous advantages, including rapid action; high
gene editing efficiency; no requirement of codon optimization and promoter selection; and reduced off-target effects,
toxicity and immune responses. [3]
Trouble shooting
Since the fundamental shortcomings of widely used viral vectors, such as the carcinogenesis, limited insertion size and immune
responses, alternative non-viral delivery systems which are more available and safer are needed. The common strategies
are based on physical approaches such as electroporation, nucleofection and microinjection. However, with the rapid
development of non-viral vectors, lipid- or polymer-based nanocarriers have great potential for CRISPR/Cas9 delivery,
especially in the delivery of RNPs. Existing non-viral vectors for in vitro RNPs delivery include arginine nanoparticles
(ArgNPs)[4], zeolitic imidazole frameworks-based CRISPR-Cas9(CC-ZIFs)[5], GO-PEG-PEI[6], and CRISPR-Gold[7]. But
these vectors have two common weaknesses, which are that these vectors themselves can neither help the RNPs target
the nucleus(since they don’t target at the nucleus) nor give provements that the RNPs enter the nucleus(since they
cannot monitor the trace of the RNPs in the cells in real time. Thus we can just suppose that they entered since
the targeted gene fragment was cleaved.
What’s our solution?
Our project focuses on the construction of a vector composed of RGD-HFBI/BODIPY, which can target the nucleus of the
targeted cell and label it by NIR dye. With help of this vector, we successfully deliver the RNPs targeting eGFP
sequence into COS7-GFP cells which firmly express eGFP protein, and the change of the fluorescence intensity before
and after the transfection process indicates the cleavage efficiency. Thus, comparation of gene-editing efficiency
of RNPs with and without RGD-HFBI/BODIPY shows that our vector can improve the cleavage efficiency of the system
without extra cytotoxicity. Moreover, the result of confocal microscopy analysis can locate the RNPs in the cells,
which confirms that the reduction of fluorescence intensity are caused by cleavage of CRISPR/Cas9 system. Our delivery
system also has potential of being applied to other gene fragments of other types of cells, for example oncogene
of Hela, which promise a chance of bright future of cancer cure.

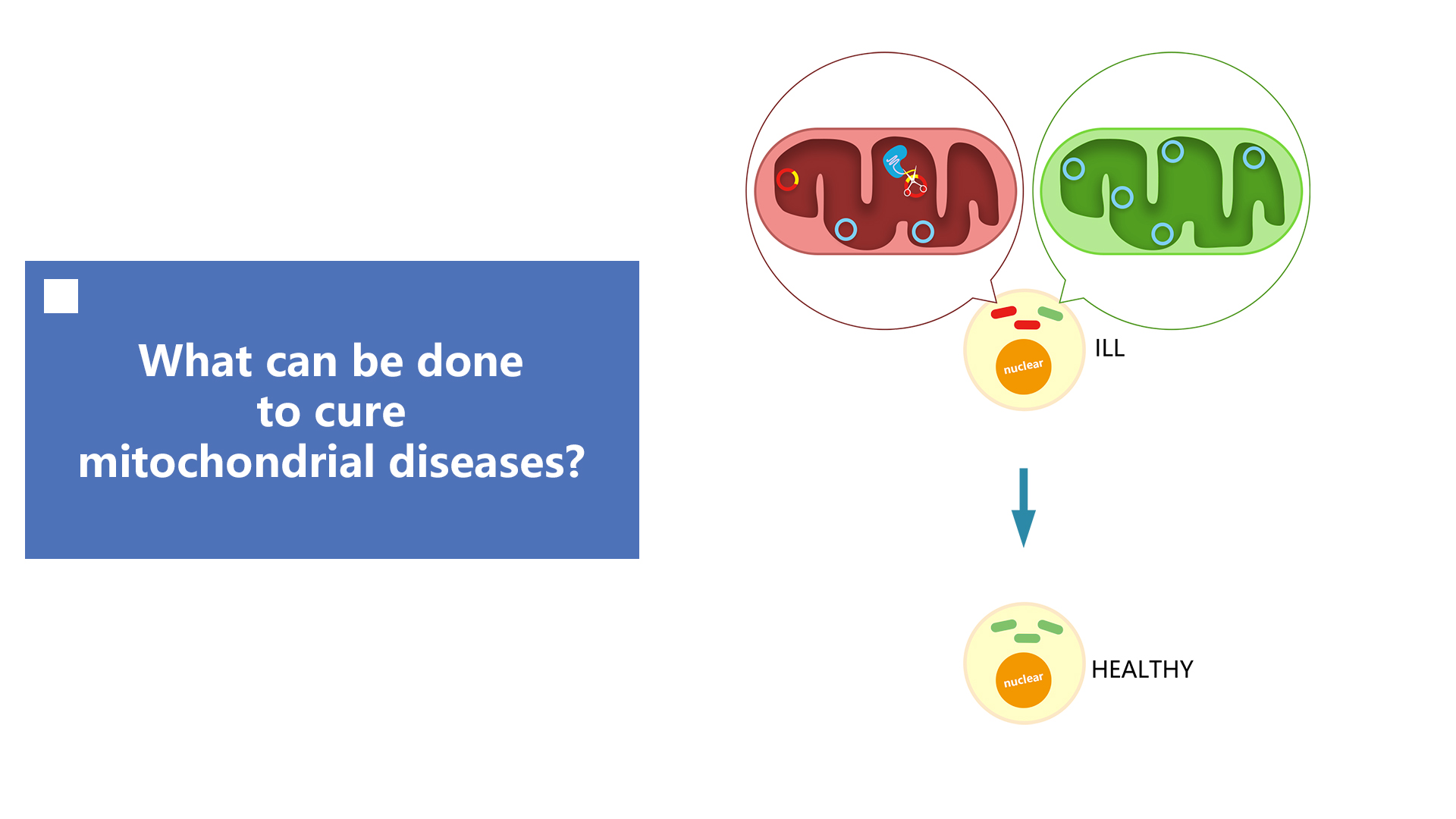
AND if that succeed,what’s NEXT?
Background
Mitochondria, the "power station" of cells, play a major role in a variety of cellular processes, including energy production,
calcium homeostasis, cell signaling, and apoptosis. Mitochondrial DNA (mtDNA) mutations primarily distribute to various
mitochondrial diseases, which have a significant impact on human neural, audiovisual and nutritional functions. However,
one method of curation, mitochondrial replacement therapy (MRT) requires DNA from different individuals, which raises
medical, safety, and ethical concerns. The other way of curation is gene edit, though TALENs and ZFNs have been used
to specifically eliminate mitochondrial disease-associated mutations of genome, the difficulties in protein design,
synthesis, and validation are still obstacles to the spread of these engineered nucleases in daily applications.
What’s our plan?
Therefore, we plan to utilise the CRISPR-Cas9 system to eliminate abnormal mutants and cure mitochondrial diseases. Compared
with the classical methods, for example, ZFN and Talen platforms that target through interactions of protein and
DNA, the CRISPR-Cas9 system uses simple base pairing rules between engineered RNA and target DNA sites. Simple programming,
unique DNA cleavage mechanisms, and the ability of multiplex target recognition enable this technique to accurately
and efficiently locate, edit, modify, regulate and mark a wide range of mitochondrial genomic loci.
Improved sensitivity of metal iron detection based on dCas9 system
Background
Human activities lead to increased environmental pollution nowadays, and since environmental pollution poses a threat
to ecosystems and human health, constant monitoring of hazardous chemicals, compounds and contaminants is needed.
Moreover, in order to successfully implement an effective bioremediation strategy, rapid detection and quantification
of pollutants is critical.
Trouble shooting
Sites suspected of being contaminated with toxic compounds are traditionally analyzed and quantified by a variety of
analytical methods including atomic absorption spectroscopy, mass spectrometry, emission spectroscopy, gas chromatography,
and variations of these methods. However, these chemical analysis methods are commonly expensive, time consuming.
Moreover, they require samples delivered from the local field to the laboratory and provide little information about
the bioavailability of metal ions. In addition, these technologies may not be available worldwide, especially in
developing countries, which limits their potential. These factors indicate the need for devices that are capable
of continuously monitoring bioavailability and in situ toxic compound concentrations in hazardous areas in real time.
What’s our solution?
We looked into the existing biosensors and found that the limit of detection of the metal ions concentration is relatively
low, so we planned to find a way to modify this circuit so that the concentration of metal ions detected by this
loop can reach lower. We then choose arsenic ions as our test object. We construct a loop for detecting arsenic ions,
which consists of the J23104 promoter, an arsenic-resistant protein, an arsenic promoter and a far-infrared fluorescent
protein. And making use of the activation function of dCas9, we can enhance the expression of fluorescence by enhancing
the transcription of the detection loop, so that the arsenic ion can be detected more clearly and its limit of detection
can be lowered. Similarly, this method can be widely applied to other metal ion detection circuits, which promise
a bright future for environmental protection.

References
[1] Chen J S, Ma E, Harrington L B, et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity.[J].
Science, 2018:eaar6245.
[2] Li S Y, Cheng Q X, Wang J M, et al. CRISPR-Cas12a-assisted nucleic acid detection.[J]. Cell Biology, 2018.
[3] Liu C, Zhang L, Liu H, et al. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications[J]. Journal of Controlled Release Official Journal of the Controlled Release Society, 2017, 266:17.
[4] Mout R, Ray M, Yesilbag T G, et al. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing.[J]. Acs Nano, 2017, 11(3):2452.
[5] Alsaiari S K, Patil S, Alyami M, et al. Endosomal Escape and Delivery of CRISPR/Cas9 Genome Editing Machinery Enabled by Nanoscale Zeolitic Imidazolate Framework[J]. Journal of the American Chemical Society, 2017, 140(1):143-146.
[6] Yue H, Zhou X, Cheng M, et al. Graphene oxide-mediated Cas9/sgRNA delivery for efficient genome editing.[J]. Nanoscale, 2017, 10(3).
[7] Lee K, Conboy M, Park H M, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair.[J]. 2017, 1(11).
[2] Li S Y, Cheng Q X, Wang J M, et al. CRISPR-Cas12a-assisted nucleic acid detection.[J]. Cell Biology, 2018.
[3] Liu C, Zhang L, Liu H, et al. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications[J]. Journal of Controlled Release Official Journal of the Controlled Release Society, 2017, 266:17.
[4] Mout R, Ray M, Yesilbag T G, et al. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing.[J]. Acs Nano, 2017, 11(3):2452.
[5] Alsaiari S K, Patil S, Alyami M, et al. Endosomal Escape and Delivery of CRISPR/Cas9 Genome Editing Machinery Enabled by Nanoscale Zeolitic Imidazolate Framework[J]. Journal of the American Chemical Society, 2017, 140(1):143-146.
[6] Yue H, Zhou X, Cheng M, et al. Graphene oxide-mediated Cas9/sgRNA delivery for efficient genome editing.[J]. Nanoscale, 2017, 10(3).
[7] Lee K, Conboy M, Park H M, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair.[J]. 2017, 1(11).