Difference between revisions of "Team:Toulouse-INSA-UPS/Design"
Gaellebordes (Talk | contribs) |
Gaellebordes (Talk | contribs) |
||
| (29 intermediate revisions by 5 users not shown) | |||
| Line 22: | Line 22: | ||
<ul class="nav nav-pills d-flex flex-row flex-wrap flex-lg-nowrap justify-content-around justify-content-lg-start" role="tablist"> | <ul class="nav nav-pills d-flex flex-row flex-wrap flex-lg-nowrap justify-content-around justify-content-lg-start" role="tablist"> | ||
<li class="nav-item"> | <li class="nav-item"> | ||
| − | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1 active" data-toggle="pill" href="#Design_Part1" role="tab">Our System</a> | + | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1 active" data-toggle="pill" href="#Design_Part1" role="tab" style="color:white !important;" >Our System</a> |
</li> | </li> | ||
<li class="nav-item"> | <li class="nav-item"> | ||
| − | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part2" role="tab">Validating Our System</a> | + | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part2" role="tab" style="color:white !important;" >Validating Our System</a> |
</li> | </li> | ||
<li class="nav-item"> | <li class="nav-item"> | ||
| − | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part3" role="tab">The Applications</a> | + | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part3" role="tab" style="color:white !important;" >The Applications</a> |
</li> | </li> | ||
<li class="nav-item"> | <li class="nav-item"> | ||
| − | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part4" role="tab">Versatility of the System</a> | + | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part4" role="tab" style="color:white !important;" >Versatility of the System</a> |
</li> | </li> | ||
<li class="nav-item"> | <li class="nav-item"> | ||
| − | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part5" role="tab"> | + | <a class="nav-link btn btn-secondary rounded-top corner-bottom mr-1" data-toggle="pill" href="#Design_Part5" role="tab" style="color:white !important;" ><i>In vivo</i> functionalisation</a> |
</li> | </li> | ||
</ul> | </ul> | ||
| Line 49: | Line 49: | ||
<hr/> | <hr/> | ||
| − | <p>To exploit the potential of cellulose as a material capable of being functionalised, we designed a three headed linker | + | <p>To exploit the potential of cellulose as a material capable of being functionalised, we designed a three-headed linker platform named Cerberus. The protein will allow us to bind a huge range of molecules to cellulose with a broad range of properties, thanks to its three fixating protein structures representing the three heads of the system.</p> |
| − | <p>We ran our tests on two different types of cellulose: a commercial Avicel was used to make colloidal particles named Regenerated Amorphous Cellulose (RAC), and bacterial cellulose produced by the bacteria <i>Gluconacetobacter hansenii</i>. Our three proteins (Sirius, Orthos and Cerberus) were designed to be produced in two chassis:<i> Escherichia coli</i> and <i>Pichia pastoris</i>. We chose to work with both a bacterium and a yeast because they both have their advantages.<i> E. coli </i>is a well-studied microorganism with a fast growth rate while <i>P. pastoris </i>is antibiotic-resistant, an ability that comes in handy when wanting to produce and antibacterial peptide, and it has a strong secretion capability. In order to validate our protein design we built a workflow, integrating <i>in silico</i>, <i>in vivo</i> and <i>in situ</i> validation steps as showed on | + | <p>We ran our tests on two different types of cellulose: a commercial Avicel was used to make colloidal particles named Regenerated Amorphous Cellulose (RAC), and bacterial cellulose produced by the bacteria <i>Gluconacetobacter hansenii</i>. Our three proteins (Sirius, Orthos and Cerberus) were designed to be produced in two chassis:<i> Escherichia coli</i> and <i>Pichia pastoris</i>. We chose to work with both a bacterium and a yeast because they both have their advantages.<i> E. coli </i>is a well-studied microorganism with a fast growth rate while <i>P. pastoris </i>is antibiotic-resistant, an ability that comes in handy when wanting to produce and antibacterial peptide, and it has a strong secretion capability. In order to validate our protein design we built a workflow, integrating <i>in silico</i>, <i>in vivo</i> and <i>in situ</i> validation steps as showed on Figure 1.</p> |
<figure class="figure" style="text-align:center;"> | <figure class="figure" style="text-align:center;"> | ||
<img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/4/44/T--Toulouse-INSA-UPS--Design--Youn--Schema1.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/4/44/T--Toulouse-INSA-UPS--Design--Youn--Schema1.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| − | <figcaption class="figure-caption"><strong>Figure 1:</strong> Overview of | + | <figcaption class="figure-caption"><i><strong>Figure 1:</strong> Overview of our experimental procedure</i></figcaption> |
</figure> | </figure> | ||
| Line 63: | Line 63: | ||
<hr/> | <hr/> | ||
<p>The first linker consists in a streptavidin head, which enables us to bind biotin and biotinylated compounds by using the strong affinity these two molecules have for each other. This interaction is in fact one of the strongest non-covalent bonds found in Nature with a dissociation constant of 10<sup>-13</sup> M, which is 10<sup>3</sup>-10<sup>6</sup> greater than any interaction between a ligand and its specific antibodies.</p> | <p>The first linker consists in a streptavidin head, which enables us to bind biotin and biotinylated compounds by using the strong affinity these two molecules have for each other. This interaction is in fact one of the strongest non-covalent bonds found in Nature with a dissociation constant of 10<sup>-13</sup> M, which is 10<sup>3</sup>-10<sup>6</sup> greater than any interaction between a ligand and its specific antibodies.</p> | ||
| − | <p>Of all the interacting pairs known and used today, we chose the biotin-streptavidin because of its appealing characteristics | + | <p>Of all the interacting pairs known and used today, we chose the biotin-streptavidin because of its appealing characteristics suitable for a whole range of applications<sup>1</sup>. The high affinity ensures that once formed the complex will not be changed by pH variations or washes, and that the binding is highly specific and will only specifically target certain molecules. Due to its small size, biotin does not alter their biological activity or other properties when attached to macromolecules. Finally, what makes streptavidin a great choice for our fusion protein is that it is very stable and its ability to bind to biotin is not easily destroyed even under harsh conditions.</p> |
| − | <p> Also, the fact that biotinylated compounds such as fluorophores, enzymes and proteins are commercially available encouraged us to use this system in our project. All these aspects also explain why the use of the streptavidin-biotin interaction was developed in the field of biotechnology. It | + | <p> Also, the fact that biotinylated compounds such as fluorophores, enzymes and proteins are commercially available encouraged us to use this system in our project. All these aspects also explain why the use of the streptavidin-biotin interaction was developed in the field of biotechnology. It allows a broad range of applications, especially in immunological and nucleic acid hybridization assays.</p> |
| − | <p>During our experiments, we decided to use two types of streptavidin: the wild-type which is tetrameric because we have data on its interaction with biotin as several iGEM teams already described the system, and | + | <p>During our experiments, we decided to use two types of streptavidin: the wild-type which is tetrameric because we have data on its interaction with biotin as several iGEM teams already described the system, and mSA2 which is monomeric and the result of a fusion of streptavidin and rhizavidin because we feared that the tetrameric streptavidin would cause aggregation of Cerberus<sup>2</sup>. Both have their pros and cons, for this reason we chose to work with both. Due to its capacity to bind four molecules of biotin per tetramer, the wild-type has an unmatched affinity for biotin. However, the monomeric version mSA2 has a shorter nucleic sequence and it is less prone to have non-specific interactions. Of all the existing non-tetrameric streptavidin, mSA2 has the highest biotin affinity (K<sub>D</sub> = 2.8 nM)<sup>3</sup>, which increases its ability to bind biotinylated compounds. Additionally, mSA2 is easier to produce as our experiments have shown, so we did not conduct all our proofs of concept with the tetrameric type.</p> |
<h3 class="heavy">Choosing the Third Head </h3><hr/> | <h3 class="heavy">Choosing the Third Head </h3><hr/> | ||
<p>The second linker of our three heads system is composed of 4-azido-L-phenylalanine (AzF), an unnatural amino acid, used to covalently bind alkyne-decorated molecules by click chemistry.</p> | <p>The second linker of our three heads system is composed of 4-azido-L-phenylalanine (AzF), an unnatural amino acid, used to covalently bind alkyne-decorated molecules by click chemistry.</p> | ||
| − | <p>At the beginning of the 21<sup>st</sup> century, the emergence of the reaction called “click chemistry” revolutionized bioconjugate chemistry. Thanks to this, coupling molecular fragments to synthesize conjugates has become easier. Following | + | <p>At the beginning of the 21<sup>st</sup> century, the emergence of the reaction called “click chemistry” revolutionized bioconjugate chemistry. Thanks to this, coupling molecular fragments to synthesize conjugates has become easier. Following this, biorthogonal reactions were developed. The most widely used is the copper-catalyzed azide-alkyne cycloaddition reaction, shortened to CuAAC<sup>4</sup>. Catalysed by Cu(I) ions, this reaction involves the azide group of our UnAA and an alkyne group. The main problem of this method is that the cytotoxicity of copper on cells limits its use in assays that take place in the cellular environment. To circumvent this problem, the strain-promoted azide−alkyne cycloaddition reaction, shortened to SPAAC, was introduced in 2004 and is used more and more nowadays. This reaction presents the advantage of not needing a catalyst, so it not toxic for the cells, and it can be implemented in physiologic pH conditions (around 7.5).</p> |
<h3 class="heavy">References</h3> | <h3 class="heavy">References</h3> | ||
<hr/> | <hr/> | ||
| Line 87: | Line 87: | ||
<h3 class="heavy">Experimental Plan: Sirius</h3> | <h3 class="heavy">Experimental Plan: Sirius</h3> | ||
<hr/> | <hr/> | ||
| − | + | <p>The first step of our experimental plan was to prove the affinity of our CBM3a to cellulose. To do that, we designed our first protein, Sirius. It was constructed by fusing CBM3a to mRFP1, which is an engineered version of DsRED but with a monomeric characteristic (Figure 1).</p> | |
| + | <div class="center"> | ||
<figure class="figure" style="text-align:center;"> | <figure class="figure" style="text-align:center;"> | ||
<img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/8/81/T--Toulouse-INSA-UPS--Design--Youn--Sirius.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/8/81/T--Toulouse-INSA-UPS--Design--Youn--Sirius.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| Line 93: | Line 94: | ||
</figure> | </figure> | ||
</div> | </div> | ||
| − | <p> | + | <p>This part was cloned into pET28 containing a pT7Lac promoter and a <i>C</i>-terminus His-tag. Once the cloning was validated by sequencing, we chose to transform it into <i>E. coli</i> BL21 DE3 because of its high protein production yield. Once produced, Sirius was purified by Immobilized Metal Affinity Chromatography (IMAC) on cobalt resin column, and the different fractions we collected were analysed on SDS-PAGE to make sure our protein was produced at the expected size. </p> |
| − | <p>Then, the binding affinity to cellulose was proved by using regenerated Avicel in a method called pull-down assay. The pull-down assay consists in separating soluble and insoluble compounds by using centrifugation. Two controls were used during the experiment: one with the Tris-HCl buffer and the Avicel, and the other with mRFP1 alone and the Avicel. These controls allow us to prove that the fluorescent protein binds to cellulose through our CBM3a and not directly. We then measured the fluorescence intensity in our tubes after several washes.</p> | + | <p>Then, the binding affinity to cellulose was proved by using regenerated Avicel in a method called pull-down assay (Figure 2). The pull-down assay consists in separating soluble and insoluble compounds by using centrifugation. Two controls were used during the experiment: one with the Tris-HCl buffer and the Avicel, and the other with mRFP1 alone and the Avicel. These controls allow us to prove that the fluorescent protein binds to cellulose through our CBM3a and not directly. We then measured the fluorescence intensity in our tubes after several washes.</p> |
| + | |||
| + | <figure class="figure" style="text-align:center;"> | ||
| + | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/7/7e/T--Toulouse-INSA-UPS--Design--Gaelle--ValidationSirius.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| + | <figcaption class="figure-caption"><i><strong>Figure 2:</strong> Experimental plan to validate the CBM3a head with our Sirius protein by using pull-down assay</i> </figcaption> | ||
| + | </figure> | ||
<h3 class="heavy">Experimental Plan: Orthos</h3> | <h3 class="heavy">Experimental Plan: Orthos</h3> | ||
<hr/> | <hr/> | ||
| − | <p>After validating the affinity of our CBM3a to cellulose, we moved on the validation of the activity of the streptavidin head with a second protein called Orthos (Figure | + | <p>After validating the affinity of our CBM3a to cellulose, we moved on the validation of the activity of the streptavidin head with a second protein called Orthos (Figure 3). Orthos is the result of the fusion of binding the molecular domain, CBM3a, and the streptavidin linker, mSA2. This part ends with an amber stop codon, preventing the His-tag from the pET28 environment from being translated.</p> |
<div class="center"> | <div class="center"> | ||
<figure class="figure" style="text-align:center;"> | <figure class="figure" style="text-align:center;"> | ||
<img style="width : 40%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/2/2f/T--Toulouse-INSA-UPS--Design--Youn--Orhtos.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | <img style="width : 40%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/2/2f/T--Toulouse-INSA-UPS--Design--Youn--Orhtos.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| − | <figcaption class="figure-caption"><i><strong>Figure | + | <figcaption class="figure-caption"><i><strong>Figure 3:</strong> Representation of our Orthos protein with its CBM3a and streptavidin heads. A TEV site (orange) is present just before the streptavidin</i> </figcaption> |
</figure> | </figure> | ||
</div> | </div> | ||
<p>Between the <i>C</i>-terminus of the streptavidin amino acid sequence and the <i>N</i>-terminus of the linker of the CBM3a, we introduced a TEV-protease site. This insertion may provide some strategy of releasing of the streptavidin and the biotinylated protein interacting with it if required, simply by incubating our complex with TEV protease.</p> | <p>Between the <i>C</i>-terminus of the streptavidin amino acid sequence and the <i>N</i>-terminus of the linker of the CBM3a, we introduced a TEV-protease site. This insertion may provide some strategy of releasing of the streptavidin and the biotinylated protein interacting with it if required, simply by incubating our complex with TEV protease.</p> | ||
| − | <p>After validating Orthos by sequencing, we produced it in <i>E. coli | + | <p>After validating Orthos by sequencing, we produced it in <i>E. coli</i> BL21 DE3 and purified it by using the cellulose affinity proven with Sirius.</p> |
| − | <p>To study its affinity for biotinylated proteins, we needed to <i>in vivo</i> biotinylate proteins. Thus, we fused mTagBFP with an Avi-tag, and cloned it in pETDuet1 which already had the sequence coding for BirA. BirA is a small protein able to recognise the 15-residues Avi-tag and to biotinylate the molecule attached to it. Once the system was transformed into <i>E. coli | + | <p>To study its affinity for biotinylated proteins, we needed to <i>in vivo</i> biotinylate proteins. Thus, we fused mTagBFP with an Avi-tag, and cloned it in pETDuet1 which already had the sequence coding for BirA. BirA is a small protein able to recognise the 15-residues Avi-tag and to biotinylate the molecule attached to it. Once the system was transformed into <i>E. coli</i> BL21 DE3 and induced with IPTG, we used the cellular lysate to prove the affinity of Orthos for biotinylated compounds by measuring the fluorescence after a pull-down assay (Figure 4). The controls used for Orthos were the same ones than for Sirius: one with the Tris-HCl buffer, and one with the biotinylated fluorescent protein.</p> |
| + | |||
| + | <figure class="figure" style="text-align:center;"> | ||
| + | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/6/67/T--Toulouse-INSA-UPS--Design--Gaelle--ValidationOrthos.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| + | <figcaption class="figure-caption"><i><strong>Figure 4:</strong> Experimental plan to validate the streptavidin head with our Orthos protein by using the CBM3a affinity for cellulose and the pull-down assay</i> </figcaption> | ||
| + | </figure> | ||
<h3 class="heavy">Experimental Plan: Cerberus</h3> | <h3 class="heavy">Experimental Plan: Cerberus</h3> | ||
<hr/> | <hr/> | ||
| − | <p>Now that two heads were validated, we still had to prove the activity of unnatural amino acid. For that, we used a third protein called Cerberus (Figure | + | <p>Now that two heads were validated, we still had to prove the activity of unnatural amino acid. For that, we used a third protein called Cerberus (Figure 5). Cerberus is a protein of 380 to 386 amino acids (depending on the type of streptavidin chosen) and 41 kDa. The affinity of the CBM3a to crystalline cellulose is around 2.9x10<sup>7</sup> M<sup>-1</sup>.<sup>1</sup></p> |
<div class="center"> | <div class="center"> | ||
<figure class="figure" style="text-align:center;"> | <figure class="figure" style="text-align:center;"> | ||
<img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/c/ca/T--Toulouse-INSA-UPS--Design--Youn--Cerberus.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/c/ca/T--Toulouse-INSA-UPS--Design--Youn--Cerberus.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| − | <figcaption class="figure-caption"><i><strong>Figure | + | <figcaption class="figure-caption"><i><strong>Figure 5:</strong> Representation of our Cerberus protein with its three binding domains, CBM3a, streptavidin and 4-azido-L-phenylalanine</i> </figcaption> |
</figure> | </figure> | ||
</div> | </div> | ||
| − | <p>The construction is similar to Orthos’ but to incorporate the UnAA, we had to hack into the genetic code. To do so, we brought | + | <p>The construction is similar to Orthos’ but to incorporate the UnAA, we had to hack into the genetic code. To do so, we brought an orthogonal aminoacyl tRNA synthase (aaRS)/tRNA pair to <i>E. coli</i>. The amber stop codon present in our construction is now recognised by the tRNA and it becomes an incorporation site for the 4-azido-L-phenylalanine (AzF).</p> |
| − | <p>This strategy was | + | <p>This strategy was conceived for specific purification because the His-tag will only be attached to AzF-containing proteins. Once validated, the part was co-transformed into <i>E. coli</i> BL21 DE3 with pEVOL-AzF, a plasmid containing tRNA and aaRS from <i>Methylococcus ianaschii</i>. We purified the production by using IMAC purification on cobalt column (Figure 6).</p> |
| − | <p> To validate the activity of the UnAA, we used fluorescein grafted with DBCO | + | <p> To validate the activity of the UnAA, we used fluorescein grafted with DBCO to perform SPAAC. This compound clicked with Cerberus’s head and after several washes we measured the difference of fluorescence between cellulose samples containing or not our Cerberus protein with the fluorophore.</p> |
| + | |||
| + | <figure class="figure" style="text-align:center;"> | ||
| + | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/6/65/T--Toulouse-INSA-UPS--Design--Gaelle--ValidationCerberus.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| + | <figcaption class="figure-caption"><i><strong>Figure 6:</strong> Experimental plan to validate the AzF head with our Cerberus protein by using pull-down assay</i> </figcaption> | ||
| + | </figure> | ||
<h3 class="heavy">References </h3> | <h3 class="heavy">References </h3> | ||
| Line 138: | Line 154: | ||
<h3 class="heavy">Paramagnetic Beads</h3> | <h3 class="heavy">Paramagnetic Beads</h3> | ||
<hr/> | <hr/> | ||
| − | <p>We first aimed to bind paramagnetic beads on the UnAA with SPAAC reaction, imagining that we could this way create magnetic cellulose that could be used as a new way to extract compounds in cells, for example, by biotinylating and binding the molecule of interest on the streptavidin head. We bought beads already attached to a DBCO function, and we put them in presence of our Cerberus under agitation overnight to allow the click reaction to occur. The next day, a pull down-assay is made, and the magnetism is observed after several washes by putting the tubes next to a magnet and by observing the functionalised cellulose moving towards it. The three controls were the Tris-HCl buffer, Cerberus alone, and the paramagnetic beads alone.</p> | + | <p>We first aimed to bind paramagnetic beads on the UnAA with SPAAC reaction, imagining that we could this way create magnetic cellulose that could be used as a new way to extract compounds in cells, for example, by biotinylating and binding the molecule of interest on the streptavidin head. We bought beads already attached to a DBCO function, and we put them in presence of our Cerberus under agitation overnight to allow the click reaction to occur. The next day, a pull down-assay is made, and the magnetism is observed after several washes by putting the tubes next to a magnet and by observing the functionalised cellulose moving towards it (Figure 1). The three controls were the Tris-HCl buffer, Cerberus alone, and the paramagnetic beads alone.</p> |
| − | <h3 class="heavy">Antibacterial Peptide, | + | <figure class="figure" style="text-align:center;"> |
| + | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/6/66/T--Toulouse-INSA-UPS--Design--Gaelle--ApplicationsParamagneticBeads.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| + | <figcaption class="figure-caption"><i><strong>Figure 1:</strong> Experimental plan for testing the paramagnetic beads on the AzF head</i> </figcaption> | ||
| + | </figure> | ||
| + | |||
| + | <h3 class="heavy">Antibacterial Peptide, scygonadin</h3> | ||
<hr/> | <hr/> | ||
| Line 146: | Line 167: | ||
<figure class="figure" style="text-align:center;"> | <figure class="figure" style="text-align:center;"> | ||
<img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/d/da/T--Toulouse-INSA-UPS--design--Youn--Scygo.jpg" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/d/da/T--Toulouse-INSA-UPS--design--Youn--Scygo.jpg" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| − | <figcaption class="figure-caption"><i><strong>Figure 2:</strong> | + | <figcaption class="figure-caption"><i><strong>Figure 2:</strong> Experimental plan for testing scygonadin on a petri dish coated with bacteria (1: scygonadin; 2: scygonadin bound to Cerberus; 3: Cerberus; 4: antibiotic)</i> </figcaption> |
</figure> | </figure> | ||
<h3 class="heavy">Testing the TEV site</h3><hr/> | <h3 class="heavy">Testing the TEV site</h3><hr/> | ||
| − | <p>This experiment is the same as the validation of our Orthos protein, except that one of the washes of the pull-down assay is replaced with a solution containing the TEV protease (see our Experiments page for further details) and that we use our Cerberus protein instead. The protein is incubated with the protease for 2 hours at 30°C. The fluorescence is then observed after centrifugation, showing that it is now present in the supernatant and not in the pellet as it used to be, showing and the biotinylated fluorescent protein was cut off from the rest of our protein. In the elution fraction, no fluorescence should be observed and measured as it was in the supernatant, and hence replaced with Tris-HCl buffer. The negative control is the biotinylated protein with cellulose.</p> | + | <p>This experiment is the same as the validation of our Orthos protein, except that one of the washes of the pull-down assay is replaced with a solution containing the TEV protease (see our Experiments page for further details) and that we use our Cerberus protein instead. The protein is incubated with the protease for 2 hours at 30°C. The fluorescence is then observed after centrifugation, showing that it is now present in the supernatant and not in the pellet as it used to be, showing and the biotinylated fluorescent protein was cut off from the rest of our protein. In the elution fraction, no fluorescence should be observed and measured as it was in the supernatant, and hence replaced with Tris-HCl buffer. The negative control is the biotinylated protein with cellulose. This test was performed at the end of our time in the lab, which explains why we did not have time to optimize the protocol after the first try.</p> |
<h3 class="heavy">Binding Two Different Molecules at Once</h3><hr/> | <h3 class="heavy">Binding Two Different Molecules at Once</h3><hr/> | ||
| − | <p> | + | |
| + | <p>Cerberus is designed to bind a wide variety of molecules, but it is also possible to bind two different molecules on its streptavidine and AzF heads. Interestingly, it is also possible to invert the function of its heads with small adapter molecules like biotin with a DBCO moeity. To illustrate both possibilities, we decided to bind the paramagnetic beads on the AzF head, then a DBCO-Biotin on the streptavidin head and finally a fluorescein-azide molecule on the newly added DBCO moeity (Figure 3).</p> | ||
| + | |||
| + | <figure class="figure" style="text-align:center;"> | ||
| + | <img style="width : 70%; heigth = auto;" src="https://static.igem.org/mediawiki/2018/e/ed/T--Toulouse-INSA-UPS--Design--Gaelle--TwoMoleculesAtOnce.png" class="figure-img img-fluid rounded" alt="A generic square placeholder image with rounded corners in a figure."> | ||
| + | <figcaption class="figure-caption"><i><strong>Figure 3:</strong> Experimental plan for binding FAM-azide on the streptavidin head and DBCO-Paramagnetic beads on the AzF head</i> </figcaption> | ||
| + | </figure> | ||
| + | |||
| + | <p>The click chemistry between the UnAA and the paramagnetic beads was made overnight, then a pull-down assay is realised to purify it. The molecules DBCO-biotin and FAM-azide are then added, and a pull-down assay is made. The magnetism is once again observed with a magnet, but this time on an UV bench so we can observe the fluorescent cellulose.</p> | ||
<h3 class="heavy">Graphene and Carbon Nanotubes</h3><hr/> | <h3 class="heavy">Graphene and Carbon Nanotubes</h3><hr/> | ||
<p>Binding graphene and carbon nanotubes to cellulose could bring interesting physical properties to it like rigidity. To bind these two compounds to our UnAA, they first needed to be activated by a reaction of diazotisation. The activation is validated by a Thermogravimetric Analysis (TGA) where the mass loss is measured to prove that the function was added in the first place. The reaction between the alkyne group and the azide function of the UnAA is still a click-chemistry but it is now copper-catalyzed (CuAAC). We only had time for the activation of the graphene and carbon nanotubes, hence we did not conduct the CuAAC reaction to functionalise our cellulose.</p> | <p>Binding graphene and carbon nanotubes to cellulose could bring interesting physical properties to it like rigidity. To bind these two compounds to our UnAA, they first needed to be activated by a reaction of diazotisation. The activation is validated by a Thermogravimetric Analysis (TGA) where the mass loss is measured to prove that the function was added in the first place. The reaction between the alkyne group and the azide function of the UnAA is still a click-chemistry but it is now copper-catalyzed (CuAAC). We only had time for the activation of the graphene and carbon nanotubes, hence we did not conduct the CuAAC reaction to functionalise our cellulose.</p> | ||
| Line 162: | Line 191: | ||
<!--PART 1 --> | <!--PART 1 --> | ||
<div class="tab-pane fade" id="Design_Part4" role="tabpanel"> | <div class="tab-pane fade" id="Design_Part4" role="tabpanel"> | ||
| − | <h2 class="heavy">Cerberus: | + | <h2 class="heavy">Cerberus: a Versatile System</h2> |
<hr/> | <hr/> | ||
| − | <h3 class="heavy">Primary Usage: | + | <h3 class="heavy">Primary Usage: Adding New Functions to Cellulose</h3> |
<hr/> | <hr/> | ||
| − | <p>Cerberus was first thought for binding | + | <p>Cerberus was first thought of for binding a variety of compounds thanks to its two very dissimilar linkers. The streptavidin linker can associate biotin and biotinylated compounds while the UnAA-containing linker will click molecules that possess an alkyne function. Using one of these heads and the CBM3a head allows to functionalise cellulose and opens the way to easily create new innovative materials.</p> |
| − | <h3 class="heavy">Secondary Usage | + | <h3 class="heavy">Secondary Usage: Combination of the Three Heads</h3> |
<hr/> | <hr/> | ||
| − | <p> | + | <p>Besides adding a function, the structure of Cerberus also permits to add two functions at the same time on cellulose (for example, magnetism and fluorescence). Such combinations expand even more the possibilities of the platform. To illustrate, imagine a fabric with Cerberus with both color and antibiotic properties: you can track if the antibiotic is still present just by assessing the color of the fabric.</p> |
| − | + | <h3 class="heavy">Third Usage: Forget About Cellulose!</h3> | |
| + | <hr/> | ||
| + | <p>The possibilities of Cerberus can go over the cellulose application field. Indeed, one could imagine applications based solely on the AzF and streptavidin heads. For example, if a bacterium has been modified to express biotin at its surface, we should be able to remove it from a culture with paramagnetic beads fixed on Cerberus' AzF head. For such applications, the CBM3a is not interesting for its cellulose binding property but for the stability it offers to the whole structure.</p> | ||
</div> | </div> | ||
| Line 177: | Line 208: | ||
<!--PART 1 --> | <!--PART 1 --> | ||
<div class="tab-pane fade" id="Design_Part5" role="tabpanel"> | <div class="tab-pane fade" id="Design_Part5" role="tabpanel"> | ||
| − | <h2 class="heavy">Functionalising Bacterial Cellulose in a | + | <h2 class="heavy">Functionalising Bacterial Cellulose in a Bioreactor</h2> |
<hr/> | <hr/> | ||
| − | <p>After validating each head separately and the ability of our platform to functionalise commercial cellulose, we had to make sure that the bacterial cellulose produced by <i>Gluconacetobacter hansenii</i> could be functionalised. To do so, we agitated a thin piece of bacterial drown in a purified fraction of Sirius and washed it several times to observe that the cellulose remains pink, demonstrating that our protein remains bound to it. The negative control used is the bacterial cellulose drown in the same fluorescent protein but without fusing it with our CBM3a. Normally, after washing this cellulose, the colour pink should not remain as the protein does not naturally bind to cellulose.</p> | + | <p>A final goal of our project was to demonstrate Cerberus' ability to functionalise cellulose <i>in vivo</i>. After validating each head separately and the ability of our platform to functionalise commercial cellulose, we had to make sure that the bacterial cellulose produced by <i>Gluconacetobacter hansenii</i> could be functionalised. To do so, we agitated a thin piece of bacterial drown in a purified fraction of Sirius and washed it several times to observe that the cellulose remains pink, demonstrating that our protein remains bound to it. The negative control used is the bacterial cellulose drown in the same fluorescent protein but without fusing it with our CBM3a. Normally, after washing this cellulose, the colour pink should not remain as the protein does not naturally bind to cellulose.</p> |
| − | <p> Once that the test is validated, we can imagine putting our chassis <i>Pichia pastoris</i> and <i>Gluconacetobacter hansenii</i> together in a | + | <p> Once that the test is validated, we can imagine putting our chassis <i>Pichia pastoris</i> and <i>Gluconacetobacter hansenii</i> together in a bioreactor, so they can respectively produce our linker with the desired function and the bacterial cellulose. Tests were run to see if the two microorganisms could grow together but due to the acidification of the medium by <i>G. hansenii</i>, the results were not conclusive. This part remains to be optimised and other experimental conditions needs to be tested. For example, as <i>P. pastoris</i> growth rate is slower than <i>G. hansenii</i>, we could test putting one after the other, and we could have a system permitting to keep the pH stable.</p> |
</div> | </div> | ||
| Line 200: | Line 231: | ||
<div id="NAV_ICON_BAR" class="nav-left-col sticky-bottom d-none d-xl-block"> | <div id="NAV_ICON_BAR" class="nav-left-col sticky-bottom d-none d-xl-block"> | ||
<ul class="nav justify-content-center"> | <ul class="nav justify-content-center"> | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
<li class="nav-item"> | <li class="nav-item"> | ||
<!--Anchor to TOP--> | <!--Anchor to TOP--> | ||
| Line 212: | Line 238: | ||
</a> | </a> | ||
</li> | </li> | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</ul> | </ul> | ||
</div> | </div> | ||
Latest revision as of 11:25, 17 October 2018
DESIGN

Our System
To exploit the potential of cellulose as a material capable of being functionalised, we designed a three-headed linker platform named Cerberus. The protein will allow us to bind a huge range of molecules to cellulose with a broad range of properties, thanks to its three fixating protein structures representing the three heads of the system.
We ran our tests on two different types of cellulose: a commercial Avicel was used to make colloidal particles named Regenerated Amorphous Cellulose (RAC), and bacterial cellulose produced by the bacteria Gluconacetobacter hansenii. Our three proteins (Sirius, Orthos and Cerberus) were designed to be produced in two chassis: Escherichia coli and Pichia pastoris. We chose to work with both a bacterium and a yeast because they both have their advantages. E. coli is a well-studied microorganism with a fast growth rate while P. pastoris is antibiotic-resistant, an ability that comes in handy when wanting to produce and antibacterial peptide, and it has a strong secretion capability. In order to validate our protein design we built a workflow, integrating in silico, in vivo and in situ validation steps as showed on Figure 1.

Choosing the First Head
The platform binds to every type of cellulose through its protein domain of the type 3 Carbohydrate Binding Module (CBM3a) family. The Imperial iGEM team from 2014 proved that of all the CBMs, it has one of the highest specificities for crystalline cellulose. Also, as it comes from the bacterium Clostridium thermocellum, it is thermostable.
Choosing the Second Head
The first linker consists in a streptavidin head, which enables us to bind biotin and biotinylated compounds by using the strong affinity these two molecules have for each other. This interaction is in fact one of the strongest non-covalent bonds found in Nature with a dissociation constant of 10-13 M, which is 103-106 greater than any interaction between a ligand and its specific antibodies.
Of all the interacting pairs known and used today, we chose the biotin-streptavidin because of its appealing characteristics suitable for a whole range of applications1. The high affinity ensures that once formed the complex will not be changed by pH variations or washes, and that the binding is highly specific and will only specifically target certain molecules. Due to its small size, biotin does not alter their biological activity or other properties when attached to macromolecules. Finally, what makes streptavidin a great choice for our fusion protein is that it is very stable and its ability to bind to biotin is not easily destroyed even under harsh conditions.
Also, the fact that biotinylated compounds such as fluorophores, enzymes and proteins are commercially available encouraged us to use this system in our project. All these aspects also explain why the use of the streptavidin-biotin interaction was developed in the field of biotechnology. It allows a broad range of applications, especially in immunological and nucleic acid hybridization assays.
During our experiments, we decided to use two types of streptavidin: the wild-type which is tetrameric because we have data on its interaction with biotin as several iGEM teams already described the system, and mSA2 which is monomeric and the result of a fusion of streptavidin and rhizavidin because we feared that the tetrameric streptavidin would cause aggregation of Cerberus2. Both have their pros and cons, for this reason we chose to work with both. Due to its capacity to bind four molecules of biotin per tetramer, the wild-type has an unmatched affinity for biotin. However, the monomeric version mSA2 has a shorter nucleic sequence and it is less prone to have non-specific interactions. Of all the existing non-tetrameric streptavidin, mSA2 has the highest biotin affinity (KD = 2.8 nM)3, which increases its ability to bind biotinylated compounds. Additionally, mSA2 is easier to produce as our experiments have shown, so we did not conduct all our proofs of concept with the tetrameric type.
Choosing the Third Head
The second linker of our three heads system is composed of 4-azido-L-phenylalanine (AzF), an unnatural amino acid, used to covalently bind alkyne-decorated molecules by click chemistry.
At the beginning of the 21st century, the emergence of the reaction called “click chemistry” revolutionized bioconjugate chemistry. Thanks to this, coupling molecular fragments to synthesize conjugates has become easier. Following this, biorthogonal reactions were developed. The most widely used is the copper-catalyzed azide-alkyne cycloaddition reaction, shortened to CuAAC4. Catalysed by Cu(I) ions, this reaction involves the azide group of our UnAA and an alkyne group. The main problem of this method is that the cytotoxicity of copper on cells limits its use in assays that take place in the cellular environment. To circumvent this problem, the strain-promoted azide−alkyne cycloaddition reaction, shortened to SPAAC, was introduced in 2004 and is used more and more nowadays. This reaction presents the advantage of not needing a catalyst, so it not toxic for the cells, and it can be implemented in physiologic pH conditions (around 7.5).
References
- Diamandis EP, Christopoulos TK: The biotin-(strept)avidin system: principles and applications in biotechnology. Clin Chem 1991, 37:625-636.
- Lim KH, Huang H, Pralle A, Park S: Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnol Bioeng 2013, 110:57-67. DOI: 10.1002/bit.24605.
- Qingqing Li QL, Xiaojian Ma, Yaping Wang, Mengjie Dong, Zhen Zhang and Lixin Ma: High-level expression of biotin ligase BirA from Escherichia coli K12 in Pichia pastoris KM71.
- Pickens CJ, Johnson SN, Pressnall MM, Leon MA, Berkland CJ: Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide-Alkyne Cycloaddition. Bioconjug Chem 2018, 29:686-701. DOI: 10.1021/acs.bioconjchem.7b00633.
Validating Our System
Validating each head of our system was the first step we had to go through, even before thinking about adding a whole range of properties. In order to do so, we designed three proteins: Sirius, Orthos and Cerberus. The aim of these proteins is to validate our system one head after the other.
Experimental Plan: Sirius
The first step of our experimental plan was to prove the affinity of our CBM3a to cellulose. To do that, we designed our first protein, Sirius. It was constructed by fusing CBM3a to mRFP1, which is an engineered version of DsRED but with a monomeric characteristic (Figure 1).

This part was cloned into pET28 containing a pT7Lac promoter and a C-terminus His-tag. Once the cloning was validated by sequencing, we chose to transform it into E. coli BL21 DE3 because of its high protein production yield. Once produced, Sirius was purified by Immobilized Metal Affinity Chromatography (IMAC) on cobalt resin column, and the different fractions we collected were analysed on SDS-PAGE to make sure our protein was produced at the expected size.
Then, the binding affinity to cellulose was proved by using regenerated Avicel in a method called pull-down assay (Figure 2). The pull-down assay consists in separating soluble and insoluble compounds by using centrifugation. Two controls were used during the experiment: one with the Tris-HCl buffer and the Avicel, and the other with mRFP1 alone and the Avicel. These controls allow us to prove that the fluorescent protein binds to cellulose through our CBM3a and not directly. We then measured the fluorescence intensity in our tubes after several washes.

Experimental Plan: Orthos
After validating the affinity of our CBM3a to cellulose, we moved on the validation of the activity of the streptavidin head with a second protein called Orthos (Figure 3). Orthos is the result of the fusion of binding the molecular domain, CBM3a, and the streptavidin linker, mSA2. This part ends with an amber stop codon, preventing the His-tag from the pET28 environment from being translated.

Between the C-terminus of the streptavidin amino acid sequence and the N-terminus of the linker of the CBM3a, we introduced a TEV-protease site. This insertion may provide some strategy of releasing of the streptavidin and the biotinylated protein interacting with it if required, simply by incubating our complex with TEV protease.
After validating Orthos by sequencing, we produced it in E. coli BL21 DE3 and purified it by using the cellulose affinity proven with Sirius.
To study its affinity for biotinylated proteins, we needed to in vivo biotinylate proteins. Thus, we fused mTagBFP with an Avi-tag, and cloned it in pETDuet1 which already had the sequence coding for BirA. BirA is a small protein able to recognise the 15-residues Avi-tag and to biotinylate the molecule attached to it. Once the system was transformed into E. coli BL21 DE3 and induced with IPTG, we used the cellular lysate to prove the affinity of Orthos for biotinylated compounds by measuring the fluorescence after a pull-down assay (Figure 4). The controls used for Orthos were the same ones than for Sirius: one with the Tris-HCl buffer, and one with the biotinylated fluorescent protein.

Experimental Plan: Cerberus
Now that two heads were validated, we still had to prove the activity of unnatural amino acid. For that, we used a third protein called Cerberus (Figure 5). Cerberus is a protein of 380 to 386 amino acids (depending on the type of streptavidin chosen) and 41 kDa. The affinity of the CBM3a to crystalline cellulose is around 2.9x107 M-1.1

The construction is similar to Orthos’ but to incorporate the UnAA, we had to hack into the genetic code. To do so, we brought an orthogonal aminoacyl tRNA synthase (aaRS)/tRNA pair to E. coli. The amber stop codon present in our construction is now recognised by the tRNA and it becomes an incorporation site for the 4-azido-L-phenylalanine (AzF).
This strategy was conceived for specific purification because the His-tag will only be attached to AzF-containing proteins. Once validated, the part was co-transformed into E. coli BL21 DE3 with pEVOL-AzF, a plasmid containing tRNA and aaRS from Methylococcus ianaschii. We purified the production by using IMAC purification on cobalt column (Figure 6).
To validate the activity of the UnAA, we used fluorescein grafted with DBCO to perform SPAAC. This compound clicked with Cerberus’s head and after several washes we measured the difference of fluorescence between cellulose samples containing or not our Cerberus protein with the fluorophore.
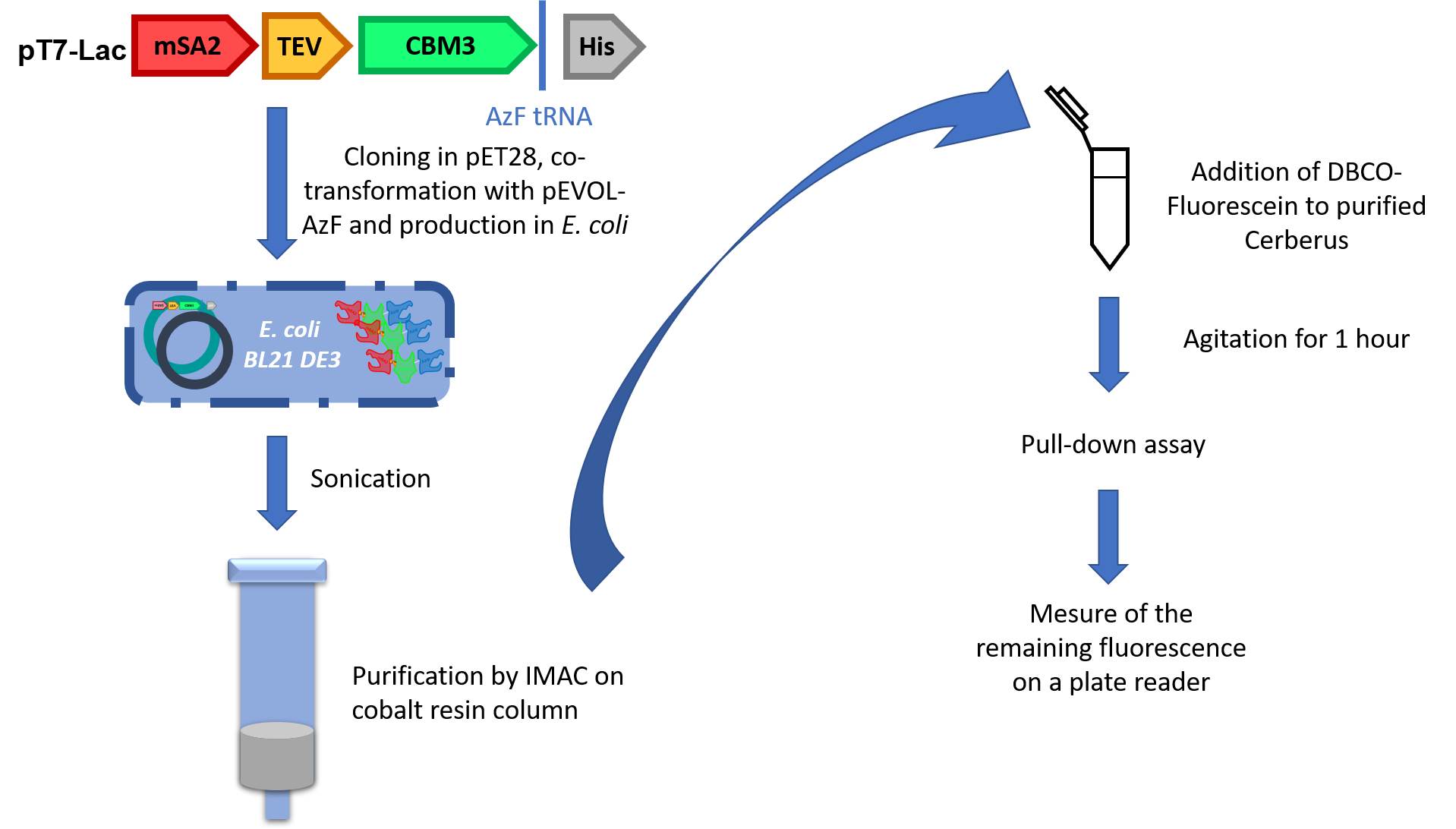
References
- Tomme P, Boraston A, McLean B, Kormos J, Creagh AL, Sturch K, Gilkes NR, Haynes CA, Warren RA, Kilburn DG: Characterization and affinity applications of cellulose-binding domains. J Chromatogr B Biomed Sci Appl 1998, 715:283-296.
The Applications of Our System
Paramagnetic Beads
We first aimed to bind paramagnetic beads on the UnAA with SPAAC reaction, imagining that we could this way create magnetic cellulose that could be used as a new way to extract compounds in cells, for example, by biotinylating and binding the molecule of interest on the streptavidin head. We bought beads already attached to a DBCO function, and we put them in presence of our Cerberus under agitation overnight to allow the click reaction to occur. The next day, a pull down-assay is made, and the magnetism is observed after several washes by putting the tubes next to a magnet and by observing the functionalised cellulose moving towards it (Figure 1). The three controls were the Tris-HCl buffer, Cerberus alone, and the paramagnetic beads alone.

Antibacterial Peptide, scygonadin
The thought of having an antibacterial bandage is very appealing and it becomes more interesting with the TEV site we added. This small site enables us to release the antibacterial molecule directly into the wound just by adding a protease. After a week of culturing Pichia pastoris engineered to produce the scygonadin, the supernatant is collected after centrifugation and put with our Cerberus overnight under agitation. A pull-down assay is realised to purify the complex Cerberus-scygonadin. Then, the sample is put on a paper disk on a Petri dish coated with bacteria. The inhibition halos are observed the next day after incubation (Figure 2). The three controls are Cerberus, scygonadin and an antibiotic.

Testing the TEV site
This experiment is the same as the validation of our Orthos protein, except that one of the washes of the pull-down assay is replaced with a solution containing the TEV protease (see our Experiments page for further details) and that we use our Cerberus protein instead. The protein is incubated with the protease for 2 hours at 30°C. The fluorescence is then observed after centrifugation, showing that it is now present in the supernatant and not in the pellet as it used to be, showing and the biotinylated fluorescent protein was cut off from the rest of our protein. In the elution fraction, no fluorescence should be observed and measured as it was in the supernatant, and hence replaced with Tris-HCl buffer. The negative control is the biotinylated protein with cellulose. This test was performed at the end of our time in the lab, which explains why we did not have time to optimize the protocol after the first try.
Binding Two Different Molecules at Once
Cerberus is designed to bind a wide variety of molecules, but it is also possible to bind two different molecules on its streptavidine and AzF heads. Interestingly, it is also possible to invert the function of its heads with small adapter molecules like biotin with a DBCO moeity. To illustrate both possibilities, we decided to bind the paramagnetic beads on the AzF head, then a DBCO-Biotin on the streptavidin head and finally a fluorescein-azide molecule on the newly added DBCO moeity (Figure 3).

The click chemistry between the UnAA and the paramagnetic beads was made overnight, then a pull-down assay is realised to purify it. The molecules DBCO-biotin and FAM-azide are then added, and a pull-down assay is made. The magnetism is once again observed with a magnet, but this time on an UV bench so we can observe the fluorescent cellulose.
Graphene and Carbon Nanotubes
Binding graphene and carbon nanotubes to cellulose could bring interesting physical properties to it like rigidity. To bind these two compounds to our UnAA, they first needed to be activated by a reaction of diazotisation. The activation is validated by a Thermogravimetric Analysis (TGA) where the mass loss is measured to prove that the function was added in the first place. The reaction between the alkyne group and the azide function of the UnAA is still a click-chemistry but it is now copper-catalyzed (CuAAC). We only had time for the activation of the graphene and carbon nanotubes, hence we did not conduct the CuAAC reaction to functionalise our cellulose.
FRET
To validate the distance between the streptavidin and the AzF heads calculated by 3D molecular modelling, we thought about using Förster resonance energy transfer (FRET). We aimed to bind fluorescein on the AzF head and biotinylated BFP on the streptavidin head. Fluorescein would have been the donor chromophore and BFP the acceptor chromophore. The distance between the two heads is around 6nm (see our Modelling page), so the FRET would have been observed as its range is usually between 1 and 10nm. Due to a lack of time, we have not been able to perform this test.
Cerberus: a Versatile System
Primary Usage: Adding New Functions to Cellulose
Cerberus was first thought of for binding a variety of compounds thanks to its two very dissimilar linkers. The streptavidin linker can associate biotin and biotinylated compounds while the UnAA-containing linker will click molecules that possess an alkyne function. Using one of these heads and the CBM3a head allows to functionalise cellulose and opens the way to easily create new innovative materials.
Secondary Usage: Combination of the Three Heads
Besides adding a function, the structure of Cerberus also permits to add two functions at the same time on cellulose (for example, magnetism and fluorescence). Such combinations expand even more the possibilities of the platform. To illustrate, imagine a fabric with Cerberus with both color and antibiotic properties: you can track if the antibiotic is still present just by assessing the color of the fabric.
Third Usage: Forget About Cellulose!
The possibilities of Cerberus can go over the cellulose application field. Indeed, one could imagine applications based solely on the AzF and streptavidin heads. For example, if a bacterium has been modified to express biotin at its surface, we should be able to remove it from a culture with paramagnetic beads fixed on Cerberus' AzF head. For such applications, the CBM3a is not interesting for its cellulose binding property but for the stability it offers to the whole structure.
Functionalising Bacterial Cellulose in a Bioreactor
A final goal of our project was to demonstrate Cerberus' ability to functionalise cellulose in vivo. After validating each head separately and the ability of our platform to functionalise commercial cellulose, we had to make sure that the bacterial cellulose produced by Gluconacetobacter hansenii could be functionalised. To do so, we agitated a thin piece of bacterial drown in a purified fraction of Sirius and washed it several times to observe that the cellulose remains pink, demonstrating that our protein remains bound to it. The negative control used is the bacterial cellulose drown in the same fluorescent protein but without fusing it with our CBM3a. Normally, after washing this cellulose, the colour pink should not remain as the protein does not naturally bind to cellulose.
Once that the test is validated, we can imagine putting our chassis Pichia pastoris and Gluconacetobacter hansenii together in a bioreactor, so they can respectively produce our linker with the desired function and the bacterial cellulose. Tests were run to see if the two microorganisms could grow together but due to the acidification of the medium by G. hansenii, the results were not conclusive. This part remains to be optimised and other experimental conditions needs to be tested. For example, as P. pastoris growth rate is slower than G. hansenii, we could test putting one after the other, and we could have a system permitting to keep the pH stable.
No dogs were harmed over the course of this iGEM project.
The whole Toulouse INSA-UPS team wants to thank our sponsors, especially:









And many more. For futher information about our sponsors, please consult our Sponsors page.
The content provided on this website is the fruit of the work of the Toulouse INSA-UPS iGEM Team. As a deliverable for the iGEM Competition, it falls under the Creative Commons Attribution 4.0. Thus, all content on this wiki is available under the Creative Commons Attribution 4.0 license (or any later version). For futher information, please consult the official website of Creative Commons.
This website was designed with Bootstrap (4.1.3). Bootstrap is a front-end library of component for html, css and javascript. It relies on both Popper and jQuery. For further information, please consult the official website of Bootstrap.