Difference between revisions of "Team:Newcastle/Results/Operon"
| Line 474: | Line 474: | ||
<img src="https://static.igem.org/mediawiki/2018/6/6b/T--Newcastle--7kbminipreps.jpeg" style="width:800px"> | <img src="https://static.igem.org/mediawiki/2018/6/6b/T--Newcastle--7kbminipreps.jpeg" style="width:800px"> | ||
<p style="text-align:center"><br>Figure 16: Agarose gel electrophoresis of miniprep for transformed colonies showing two 7kb bands at position 9 on the first gel and 4 on the second. These were both from plate 4 of the 1-4 Gibson assemblies with amplified and un-amplified gBlocks.</p> | <p style="text-align:center"><br>Figure 16: Agarose gel electrophoresis of miniprep for transformed colonies showing two 7kb bands at position 9 on the first gel and 4 on the second. These were both from plate 4 of the 1-4 Gibson assemblies with amplified and un-amplified gBlocks.</p> | ||
| + | <p class="about-para">The two colonies displaying a 7kb band were sent for sequencing using primers Gb1F, Gb1R, Gb2F, Gb2R, Gb3F and Gb2R and the miniprepped product. These primers were selected as they allowed around 1kb of the gBlock to be sequenced in each tube by GATC sequencing. The primers either inconclusively or incorrectly aligned to the naringenin operon for both of these sequenced colonies. One of the colonies sequenced appeared as though the naringenin operon gBlocks had been transformed, however due to gaps in the sequence read this was not conclusive evidence of naringenin operon transformation of the cells (Figure 17). This could be due to the sensitive nature of the primers when annealing to the sequence. Overall the lack of transformation efficiency of the full 7kb operon was likely due to the size of the operon being too large for the cells to take up, so those cells transformed only took up the plasmid as this was an easier alternative.</p> | ||
</div> | </div> | ||
</div> | </div> | ||
Revision as of 21:02, 17 October 2018

Alternative Roots
Naringenin Operon Assembly Results
Results
Introduction
Guided by the successful chemotaxis results, proving that 50µM naringenin attracts the nitrogen fixing bacteria A. brasilense and H. seropedicae, we aimed to engineer a naturally colonising endophyte Pseudomonas sp. (CT 364) to produce naringenin. For proof of concept the production of naringenin would first need to be demonstrated in E. coli before being tested in Pseudomonas sp., our final chassis organism.
Naringenin biosynthesis is achieved through the expression of an operon containing four genes encoding the enzymes that constitute the naringenin biosynthetic pathway (Figure 1). This operon was previously assembled and submitted to the iGEM registry by TU Darmstadt 2014 iGEM team BBa_K1497016 and is a composite of the following four genes, each with the strong RBS (BBa_B0034):
- 4-Coumaryl ligase - 4CL BBa_K1033001
- Tyrosine ammonia lyase - TAL BBa_K1033000
- Chalcone isomerase - CHI BBa_K1497000
- Chalcone synthase - CHS BBa_K1497001
We decided to optimise the sequence for the enzyme tyrosine ammonia lyase by reducing the G/C content so that it could be synthesised by IDT BBa_K2797014. The entire operon was synthesised in four separate parts referred to as gBlocks that were subsequently used for Gibson Assembly, as this was how TU Darmstadt assembled their operon successfully. Moreover this assembly does not leave restriction site scars due to the overlapping fragments. Instead of using Pseudomonas sp. it was deemed logical to use E. coli DH5α during the cloning experiments as we had a greater understanding of its transformation. Using this as a proof of concept would allow us to carry out a preliminary screening for naringenin production before attempting to transform our endophyte.
Alongside this, in an attempt to optimise naringenin production, a new design of the naringenin operon was made in Benchling. This was based on the pathway modelling results and was constructed to show how BG28 and BG51 dual E. coli-Pseudomonas promoters with a 10-fold difference in strength could increase naringenin production.

Figure 1: The naringenin synthesis pathway from L-tyrosine.
Results
Operon and Plasmid Design
Plasmid Design
The initial plasmid design for naringenin biosynthesis was based on TU Darmstadt’s design but with a codon optimised tyrosine ammonia lyase. We used pSB1C3 as a backbone, into which we would clone each of the four necessary genes downstream of a strong ribosome binding site (BBa_B0034). This construct was under the control of a strong Anderson promoter (J23100) to allow for constitutive expression of the operon (Figure 2). Once biosynthesis under the control of J23100 is achieved, future experiments will test this under the strong constitutive T7 promoter in E. coli (Figure 3). Parts for biosynthesis in root-colonising Pseudomonas sp., will be implemented into a plasmid backbone more suitable for its uptake.
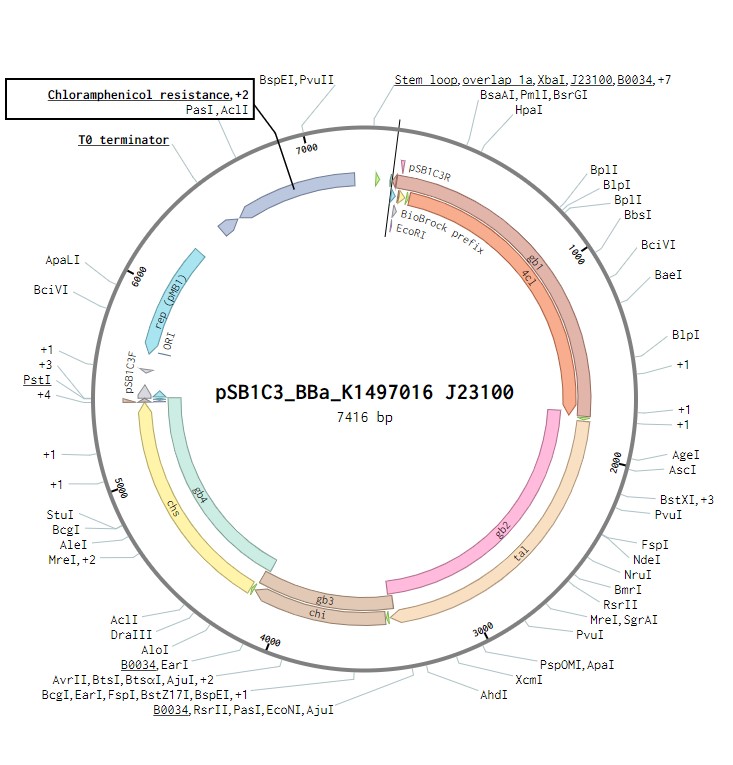
Figure 2:The naringenin biosynthetic operon under control of a J23100 promoter created in Benchling.

Figure 3:The naringenin biosynthetic operon under control of a T7 promoter created in Benchling.

Figure 4:The naringenin biosynthetic operon construct under control of a J23100 promoter created in SBOL.

Figure 5: The naringenin biosynthetic operon contruct under control of a T7 promoter created in SBOL.
Naringenin pathway modelling influenced design
Results of the naringenin pathway modelling demonstrated that weaker expression of the first two genes and 10-fold stronger expression of the last two genes would reduce the build-up of malonyl CoA and optimise naringenin synthesis. As a result of this, two synthetic promoters (BG28 and BG51) (Figure 7) were selected and an additional E. coli his operon terminator that was placed after the first two genes (Figure 6). These changes to the operon design could allow enhanced naringenin production in future experiments. More information about the pathway can be found here.

Figure 6:The naringenin biosynthetic operon under control of synthetic promoters BG28 and BG51 created in Benchling.

Figure 7: A close up of the synthetic promoters BG28 and BG51 placement in the operon, created in Benchling.

Figure 8: The naringenin biosynthetic operon contruct under control of a BG28 and BG51 promoters and an additional his operon terminator created in SBOL.
Primers were designed for amplification of the backbone and the 4 gBlocks (Table 1). These were designed in Benchling. The primers were used to prevent running out of the gBlocks synthesised by IDT and to detect the specific genes from the operon that could be present in transformed cells.
Table 1: Primers designed in Benchling for amplification of the 4 gblocks and the pSB1C3 backbone.
| Primer name | Sequence | Tm | Ta Q5 | Amplified product(bp) | Shown to work | Description |
|---|---|---|---|---|---|---|
| pSB1C3F | tactagtagcggccgctgc | 70 | 71 | 2070 | 6/8/18 | To amplify pSB1C3 backbone |
| pSB1C3R | ctctagaagcggccgcga | 70 | 71 | 2070 | 6/8/18 | To amplify pSB1C3 backbone |
| 4CLF | ccaaatcgccgccaattttc | 59 | 56 | 1686 | 6/9/18 | To amplify 4CL part |
| 4CLR | cgtcgtcgttttgaagtggt | 59.07 | 56 | 1686 | 28/9/18 | To amplify 4CL part |
| TALF | gaatgtccgaacgctacagg | 58.72 | 55 | 1649 | 28/9/18 | To amplify TAL part |
| TALR | tcggaattgagcaggtcgat | 59.18 | 56 | 1649 | 28/9/18 | To amplify TAL part |
| CHIF | ctgggcatagaggtctggag | 58.95 | 56 | 726 | 28/9/18 | To amplify CHI part |
| CHIR | caccttctccgagtactgct | 58.82 | 56 | 726 | 28/9/18 | To amplify CHI part |
| CHSF | aagacgtgcctgggttgata | 59.02 | 56 | 1197 | 28/9/18 | To amplify CHS part |
| CHSR | gcttctcctccttcaaccct | 59.01 | 56 | 1197 | 6/9/18 | To amplify CHS part |
| gb1F | ctggaattcgcggccgct | 72 | 54 | 1686 | 20/9/18 | To amplify 4CL part |
| gb1R | ttacaatccatttgctag | 53 | 54 | 1686 | 20/9/18 | To amplify 4CL part |
| gb2F | ggcaaaactagcaaatgg | 59 | 59 | 1649 | 20/9/18 | To amplify TAL part |
| gb2R | ttatcagacgggagattg | 58 | 59 | 1649 | 20/9/18 | To amplify TAL part |
| gb3F | cttgcagcaatctcccgt | 65 | 59 | 726 | 20/9/18 | To amplify CHI part |
| gb3R | ctagactccaatcactgg | 58 | 59 | 726 | 20/9/18 | To amplify CHI part |
| gb4F | tactattccagtgattgg | 54 | 55 | 1197 | 20/9/18 | To amplify CHS part |
| gb4R | cggactgcagcggccgct | 78 | 55 | 1197 | 20/9/18 | To amplify CHS part |
Results
Experimental Work
Backbone amplification
The pSB1C3 backbone was amplified, purified and quantified in preparation for Gibson assembly of the naringenin operon. Amplification was performed by PCR using Q5 ® High-Fidelity DNA Polymerase and primers pSB1C3F and pSB1C3R. The resultant PCR product was analysed by agarose gel electrophoresis and showed a band at 2kb that corresponded to the size of the plasmid. The backbone was then purified using Qiagen QIAquick PCR Purification Kit. The DNA concentration was quantified using a Qubit fluorometer and was determined to be 3.58 µg/ml. As this concentration was far too low for Gibson assembly the backbone was amplified and purified again using the same techniques resulting in a brighter band at 2kb than before (Figure 9) and a stock concentration of 26.2 µg/ml.
Protocol Details found here
Figure 9: Agarose gel showing the 2 kb band representing the amplified plasmid backbone, Bioline HyperLadderTM 1 kb is shown that was used for subsequent gel analysis.
Gibson Assemblies
A positive control of the Gibson assembly was conducted. The NEBuilder® HiFi DNA Assembly Cloning Kit was used. A positive control reagent supplied with the kit contained two overlapping dsDNA fragments and the pUC19 plasmid. The positive control was conducted to check the assembly mix was working, the protocols for transformation were correct and that the cells prepared previously were competent.
The results of the positive control showed that the E. coli DH5α cells had successfully been made competent as they were able to take up the three-part assembly of the two overlapping fragments and the pUC19 backbone (Figure 10). Furthermore the reagents and protocols used in the Gibson assemblies were shown to have worked proving their viability for use in future experiments. No colonies grew on the negative control plates (Figure 10), verifying that E. coli DH5α cells are not ordinarily resistant to amplicillin and that no contamination had occurred.

Figure 10: LB ampicillin positive control plate containing colonies from transformed cells and negative control plates with un-transformed DH5α cells.
Initial Gibson Assemblies of the naringenin operon
All Gibson assemblies were carried out using the 4 gBlocks, the pSB1C3 backbone and the competent cells induced on the 10/8/18. Gibson assemblies were conducted using the NEBuilder HiFi Assembly mix. For the assembly of 4 – 6 fragments the total concentration of DNA for the reaction had to be between 0.2 – 0.5 pmol with equimolar concentrations between all the inserts and the vector (0.05 pmol each) (Table 2). This reaction was incubated for 60 minutes at 50°C. The DH5α cells were then heat-shock transformed with the Gibson assembly product. The cells were spread onto LB plates containing chloramphenicol. Negative control plates containing un- transformed DH5α cells with no DNA were produced, to verify that DH5α cells do not have chloramphenicol resistance and that no contamination of plates occurred.


Figure 11: A) E. coli DH5α colony from cells transformed with the second Gibson Assembly. B) E. coli DH5α colony from cells transformed with the third Gibson assembly. C) Negative control plate of un- transformed E. coli DH5α cells showing no colonies.
For the first Gibson assembly (Table 2), the backbone had a very low concentration, resulting in a low transformation efficiency. There was no colony growth on the transformed plates indicating the plasmid had not been taken up. Additionally there was no colony growth on the negative control.
Following the lack of colonies, the amplified backbone with the higher concentration of 26.2µg/ml was used coupled with reducing the total reaction volume in Gibson assembly from 36µl to 20µl (Table 3). Reducing the reaction volume allowed the use of less master mix in the reaction. This method yielded two colonies from cells that had been transformed (Figure 11A and 11B) and a corresponding negative control plate (Figure 11C). It was discovered through agarose gel electrophoresis of the miniprep of these colonies that they had only taken up the 2kb plasmid without the operon, as the only bright band was at 2kb when compared to the ladder (Figure 12). The absence of colony growth indicated a low transformation efficiency, so commercial cells were used in future experiments instead of homemade competent cells.

Figure 12: Agarose gel electrophoresis of the two miniprepped colonies from the second and third Gibson assemblies. 6 columns as split the 2 colonies into 3 separate tubes for the miniprep.
Gibson Assembly transformations into commercial cells
Due to multiple failed Gibson assembly attempts the gBlocks and backbone were amplified by PCR using Q5 ® High-Fidelity DNA Polymerase. Gel extraction was performed using the QIAquick gel extraction kit. Four Gibson assemblies from combinations of these amplified, gel extracted and original un- amplified gBlocks were set up (Table 4, Table 5, Table 6 and Table 7). These were then transformed into commercial DH5α cells competent cells. As a result, 30 to 60 colonies per transformation plate grew (Figure 13).





Figure 13: Colonies from transformed cells with Gibson assemblies 1-4 using different volumes of gBlocks.
As primers had been designed for detection of the operon from transformants, colony PCR was conducted on 5 of the colonies from the plates 1-4 using Q5 ® High-Fidelity DNA Polymerase to detect the 5kb band corresponding to the full length of the operon and each individual gene. There was a lack of visible bands when gel electrophoresis was performed (Figure 14).

Figure 14: Colony PCR of colonies from transformed cells using the primers for the beginning and end of the operon and each gene.
No visible bands were present for either the 5kb operon or 2kb backbone on the gel, therefore colony PCR was not utilised in the future for the detection of the operon as the primers did not bind to any of the DNA. Therefore the colonies from future transformants would be miniprepped and agarose gel electrophoresis performed to visualise the plasmid. 45 colonies of DH5α transformants were inoculated in LB media containing chloramphenicol and those cultures were used to carry out plasmid extractions that where later run on an agarose gel (Figure 15 and Figure 16). Two colonies from plate 4 (Figure 13) showed a promising 7kb band corresponding to the full length of the operon and the plasmid.

Figure 15: Agarose gel electrophoresis of colonies from plates of DH5α transformed cells from the Gibson assemblies 1-4 with the amplified and un-amplified gBlocks, showing 2kb bands corresponding to the plasmid.

Figure 16: Agarose gel electrophoresis of miniprep for transformed colonies showing two 7kb bands at position 9 on the first gel and 4 on the second. These were both from plate 4 of the 1-4 Gibson assemblies with amplified and un-amplified gBlocks.
The two colonies displaying a 7kb band were sent for sequencing using primers Gb1F, Gb1R, Gb2F, Gb2R, Gb3F and Gb2R and the miniprepped product. These primers were selected as they allowed around 1kb of the gBlock to be sequenced in each tube by GATC sequencing. The primers either inconclusively or incorrectly aligned to the naringenin operon for both of these sequenced colonies. One of the colonies sequenced appeared as though the naringenin operon gBlocks had been transformed, however due to gaps in the sequence read this was not conclusive evidence of naringenin operon transformation of the cells (Figure 17). This could be due to the sensitive nature of the primers when annealing to the sequence. Overall the lack of transformation efficiency of the full 7kb operon was likely due to the size of the operon being too large for the cells to take up, so those cells transformed only took up the plasmid as this was an easier alternative.
Results
Conclusions
Two colonies resulting from transformed cells with the naringenin operon resulted in a 7kb band, corresponding to the size of the operon and plasmid backbone. However, sequencing was inconclusive for the first colony and incorrect for the second therefore this operon could not be classified as a working part. Future experimentation should attempt to assemble the gBlocks one by one into the plasmid backbone and gain sequencing results that show full alignment. Following this, BL21 expression cells should be transformed with the 7kb plasmid to produce naringenin. This would be extracted using ethyl acetate and measured through HPLC. HPLC of stock naringenin should be conducted for comparison, using a range of concentrations to produce a standard curve. Observing the expression of the operon using T7 promoters instead of the J23100 constitutive promoter, to see if naringenin production is enhanced. This could be implemented using T7 primers and Q5 site directed mutagenesis in E. coli. Using the BG51 and BG28 promoters this construct should be assembled into the chassis organism Pseudomonas sp.
References & Attributions
Attributions: Heather Bottomley and Patrycja Ubysz