Difference between revisions of "Team:SCAU-China/Model"
LeeKartoon (Talk | contribs) |
LeeKartoon (Talk | contribs) |
||
| Line 388: | Line 388: | ||
<span class="caret"></span> | <span class="caret"></span> | ||
<ul class='dropdown-menu'> | <ul class='dropdown-menu'> | ||
| − | + | <li><a href="https://2018.igem.org/Team:SCAU-China/ProjectOverview">Overview</a></li> | |
<li><a href="https://2018.igem.org/Team:SCAU-China/Background">Background</a></li> | <li><a href="https://2018.igem.org/Team:SCAU-China/Background">Background</a></li> | ||
<li><a href="https://2018.igem.org/Team:SCAU-China/Design">Design</a></li> | <li><a href="https://2018.igem.org/Team:SCAU-China/Design">Design</a></li> | ||
Revision as of 07:30, 16 October 2018
Abstract:
In order to increase the production of bacterial cellulose in cynobacteria, we want to base on known regulatory and metabolic information and new knowledge generated by gene expression profile. We developed a statistic model based on partial correlation and Hierarchical Clustering called HAWNA(Hierarchical And Weighted Network Analysis), to infer genetic regulatory network and gene coexpression module. We perform simulation test and combine known information of genetic regulatory pathway of PCC6803 to show that our model perform better than existing model and can enrich functional pathway significantly. We then performed our model on public microarray dataset of PCC6803 to predict new genes may influence the production of bacterial cellulose and used crisper-cas9 to knock out these genes to further prove our model. HAWNA is consisted of two analysis step
1. Causality network analysis step: We use singular vector decomposition and partial correlation analysis to find alternative genes which not annoted on our target pathway have direct correlation with our annoted pathway genes. The direct correlation means the correlation of two variables while not controlling for a third or more other variables.
2. Gene coexpression module detect analysis: We use correlation matrix and a variety of cluster algorithms to find network community and quantify the effect of genes to the whole system.
Overview:
In our result, we have demonstrated that we have developed a PCC6803 strain capable of expressing bacterial cellulose. However, we find that our strain cannot produce sufficient bacterial cellulose. To improve the production, the metabolic engineering is necessary.
Advances in metabolic engineering have enabled the modification of biosynthetic pathways in microorganisms, one major step in metabolic engineering is to overexpression or knockdown a heterologous enzyms to boost the yield of an intermediates(s) or the desired product directly. However, the genetic modification will bring two major problems in metabolic engineering:
1. The modification may result in accumulation of intermedias with no change or even decrease of desired product. Because of complexity and heterogeneity of genetic regulatory network.
2. The modification may result in potential metabolic burdens which may influent the growth of cynobacteria.

Figure1 the bacterial cellulose biosynthetic pathways,
This present a issue that which genes need to be knock out, In cellulose biosynthetic pathway(Fig 1), we can found that gene slr0897(Cyanobase access- ion number)encode endo-1,4-beta-glucanase like protein(SsGlc), and present function of cellulose degradation and salt stress tolerance. For the point2, it may not suitable to knock out this gene directly. In order to address above two problems. We decided to develop a mathematical model of gene regulatory network, to detect new genes may have functional involvement in cellulose production and measure each gene’s impact to the whole biological regulatory network.
Genetic regulatory networks(GRNs) are collections of gene-gene regulatory relations in a genome and are models that display causal relationships between gene activities. Inference of GRNs based on gene expression profile is referred to as ‘reverse engineering’, as the gene expression profile is the outcome of GRNs. Mathematically, reverse engineering is a traditional inverse problem. But GRN is really complicated by the enormously large scale of the unknowns, the inherent noisy like experimental defects and many other hidden variable my play a role in the variation of your data.
In mathmatically, we have a variety of method to inference and simulate GRNs. Most iGEM teams tend use Flux Balance Analysis to Ordinary Differential Equantion. But this way may bring two shortcomings, the first one is modelers often need an important background knowledge of your organism, and most ODEs model have a strong assumption about network structure. Second, ODEs perform not so good in a noisy gene expression profile, and may lead to overfitting problem even fit perfectly with your observed data. Compare with ODEs, Using probability graphic model seems are more better choice in large scale GRN inference, their learning techniques have a solid basis in statistics, allowing them to deal with the stochastic aspects of gene expressions and noisy measurements in a natural way, and give us more direct and easy way to draw the graph of whole GRN.
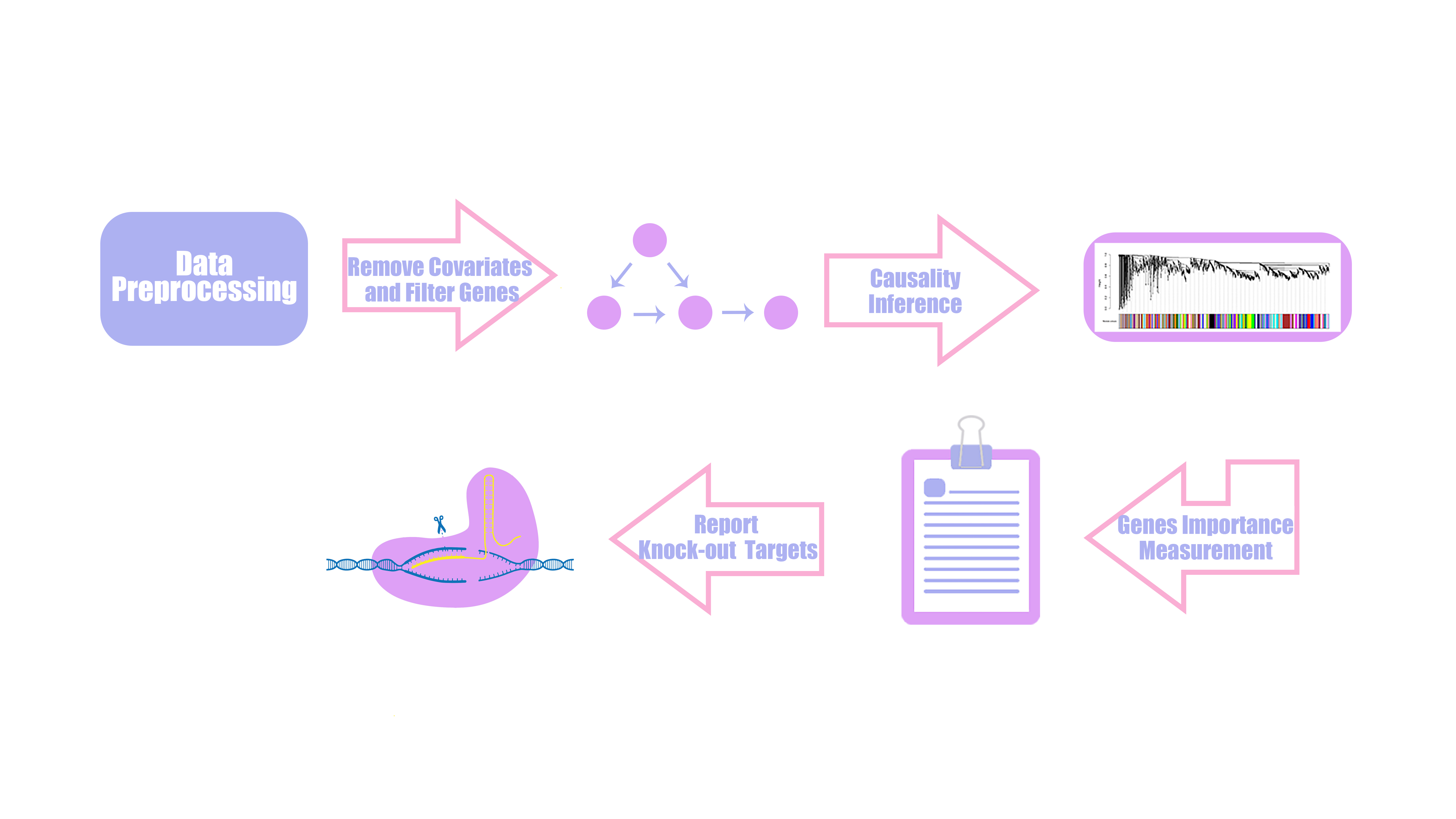
Fig2. the structure of HAWNA
Collecting and processing data
The HAWNA is based on the gene expression profile, We download the microarray data of PCC6803, The gene expression in Synechocystis sp. PCC 6803 was analyzed following culture at different light conditions(30, 50, 300, 1000, 1100, and 1300 μmol m-2 s-1. Three independent experiments were performed under each light intensity). To remove the bias of light intensity, we use linear regression model to caculate the residule variation of each gene after regressing out the light effect.
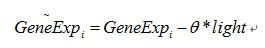
The θ is learned through OLS(ordinary least square) methods, after regressing process. We rescale the data, and use Kolmogorov-Smirnov test to test each gene’s gaussianity. This step we remove 182 genes to prevent their effects from network construction.
Partial network construction
Difference Between Causality And Correlation?
Correlation networks are widely used to explore and visualize high-dimentinal data, Constructing correlation network is really simple and include two steps: 1) the computation of all pairwise correlations for the investigated variables. And 2) a thresholding or filtering procedure to conduct significant correlation. But correlation will reach limit that it not represent causuality, For instance, we can simulate three variable. X, Y, Z
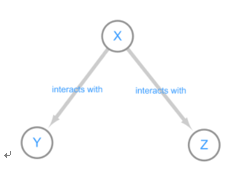
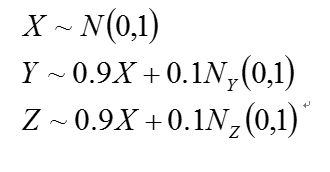
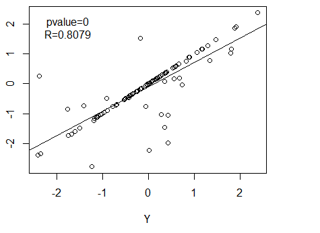
Figure 3: interact diagram of two genes(Y,Z) co-regulated(X) model
The distribution of X, Y, Z are independent, as we can see, the variable Z and Y present very strong correlation, But Y and Z is regulated by variable X, which means they are not causality correlation. If we knock out gene Y, it seems will not down-regulated the expression value of Z.
So consider the conditional correlation is really important, it can help us to remove the influence of intermedia variable, and omit some meaningless correlation.
Partial correlation model
The partical correlation is the correlation that remains between the two variables if the effect of the other variables has been regressed away. As proved in Gaussian Graphic Models theory, that the partial correlation(also called conditinal correlation) of Xa and Xb given {Xc:c≠a,b} is given by
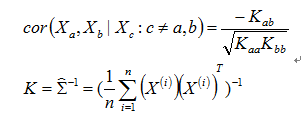
K matrix means the inverse matrix of covariance matrix. For n>p-2, we have
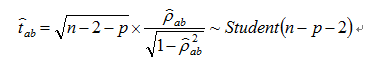
For each a and b, we can caculate the p-value of 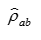 with the test statistic
with the test statistic 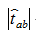
However, for most gene expression matrix, the sample size(n) is much more smaller than the number of variables(p>>n). Which will bring two problems:
1. The emprical covariance matrix is singular, and the estimation of corresponding inverse matrix(K) will be unstable.
2. The degree of freedom of test statistic will be negative.
Consider above problems, we propose an novel computational framework, based on matrix decomposition and random matrix theory, we can de-dimentional efficiently, and make accurate inference of causality network.
Information compression
To solve the problems of “curse of dimensionality” in partial correlation analysis step. It is known that most information of matrix can be compressed through a wide of different algorithms like PCA, ICA and so on. We use SVD to help us compress the information to reduce dimensions, make matrix invertible and caculate the significants of partial correlation of two genes.
The Singular Vector Decomposition(SVD) is a matrix decomposition that is very useful in many fields of applied mathmatics.For any n*p matrix A, the matrices ATA and AAT are symmetric positive semidefinite.
Any matrix A of rank r can be decomposed as
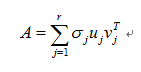
where
- r=rank(A)
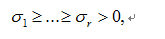
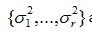 are the nonzero eigenvalues of ATA(which are also the nonzero eigenvalues of AAT), and
are the nonzero eigenvalues of ATA(which are also the nonzero eigenvalues of AAT), and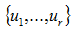 and
and
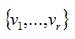 are two orthonormal families of Rn and Rn, such that
are two orthonormal families of Rn and Rn, such that
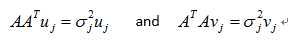
U vectors are said to be left-singular vectors, represent the characteristic vector of AAT, the range of it and the range of A coincide. So we can use left-singular vector to surragate the variation of Xc. Which means we can use a part of left-singular vectors to control the rest genes. The number of dimensions is choosen through random matrix theory(RMT), see text1 for more details of dimensions choose.
Candidate targets detection
Based above theory, In causality network inference step, We first seperate genes into two sets. The target pathway gene set and The non-target pathway gene set. For each gene on the target pathway, we caculate its’ patial correlation with all genes in non-target pathway gene set. Than, we defined the genes in non-target pathway gene set only present one interaction with the gene in target pathway gene set as candidate target.
In our analysis, We want to search the non-target pathway genes which only present positive interaction with slr0897. The psuedo code of causality network inference step is detailed below:
Pvalue = pvalue of partial correlation matrix, number of row equal to the number of target pathway genes, number of column equal to the number of non-target pathway genes
Cor = partial correlation matrix, number of row equal to the number of target pathway genes, number of column equal to the number of non-target pathway genes
for(i in 1: number of target pathway genes){
for(j in 1:number of non-target pathway genes){
#sepearate nonPathwayMatrix into two components
SVD_nonpathway <- t(nonPathwayMatrix[-j,]); # extract genej to caculate correlation, SVD_nonpathway is the rest gene expression profile which need to be controled
SVD_nonpathway <- svd(SVD_nonpathway); # SVD step
U <- SVD_nonpathway$u[,1:svdDim] # choose significant dimensions of U matrix, svdDim is determined through RMT theory
final_nonPathwayMatrix <- rbind(t(U),as.matrix(nonPathwayMatrix[j,]))
M <- rbind(final_nonPathwayMatrix,as.matrix(pathwayMatrix[i,]))
M <- t(M)
M <- as.data.frame(M) # merge genei , genej and compressed matrix U(the information of controled gene expression matrix)
test <- pcor(M) # caculate partial correlation matrix
est <- test$estimate # partial correlation matrix
P <- test$p.value # pvalue of partial correlation matrix
estimate <- as.matrix(est)
pvalue <- as.matrix(P)
Pvalue[i,j] <- pvalue[svdDim+1,svdDim+2] # input the partial correlation of genei and genej
Cor[i,j] <- estimate[svdDim+1,svdDim+2] # input the partial correlation pvalue of genei and genej
}
}
FDR control of Pvalue matrix
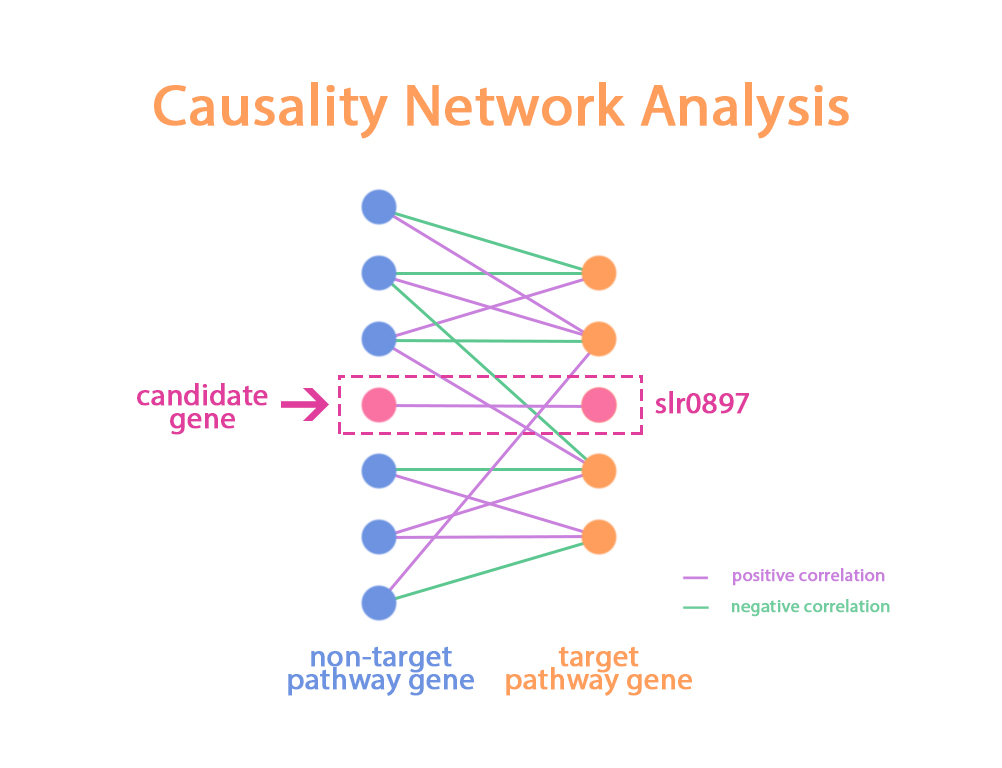
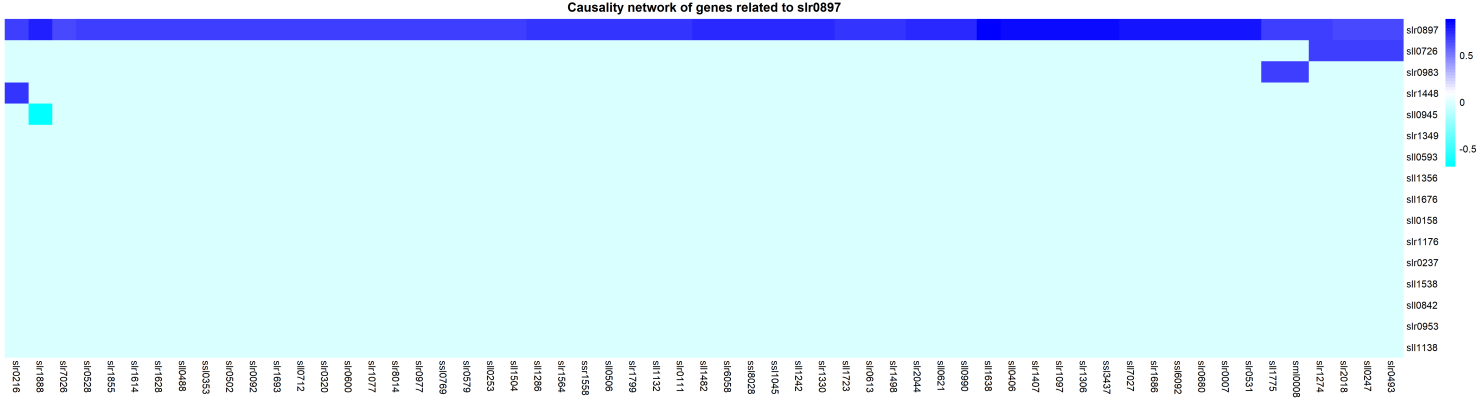
Figure 4 the heatmap of non-target pathway genes which positive correlated with slr0897
According to our result, We found that around 59 genes that positive correlate(partial correlation with slr0897)with slr0897, Besides, there are 51 genes only correlate with slr0897 across genes in target pathway.
Another question we need to address is to determine which gene is suitable to be knock out in these 51 genes. The nature of scale free property of genetic regulatory network decide that genes have different impact to the whole biological system. Motivited by this idea, the next step of HAWNA is to define the “importance score” for each target gene.
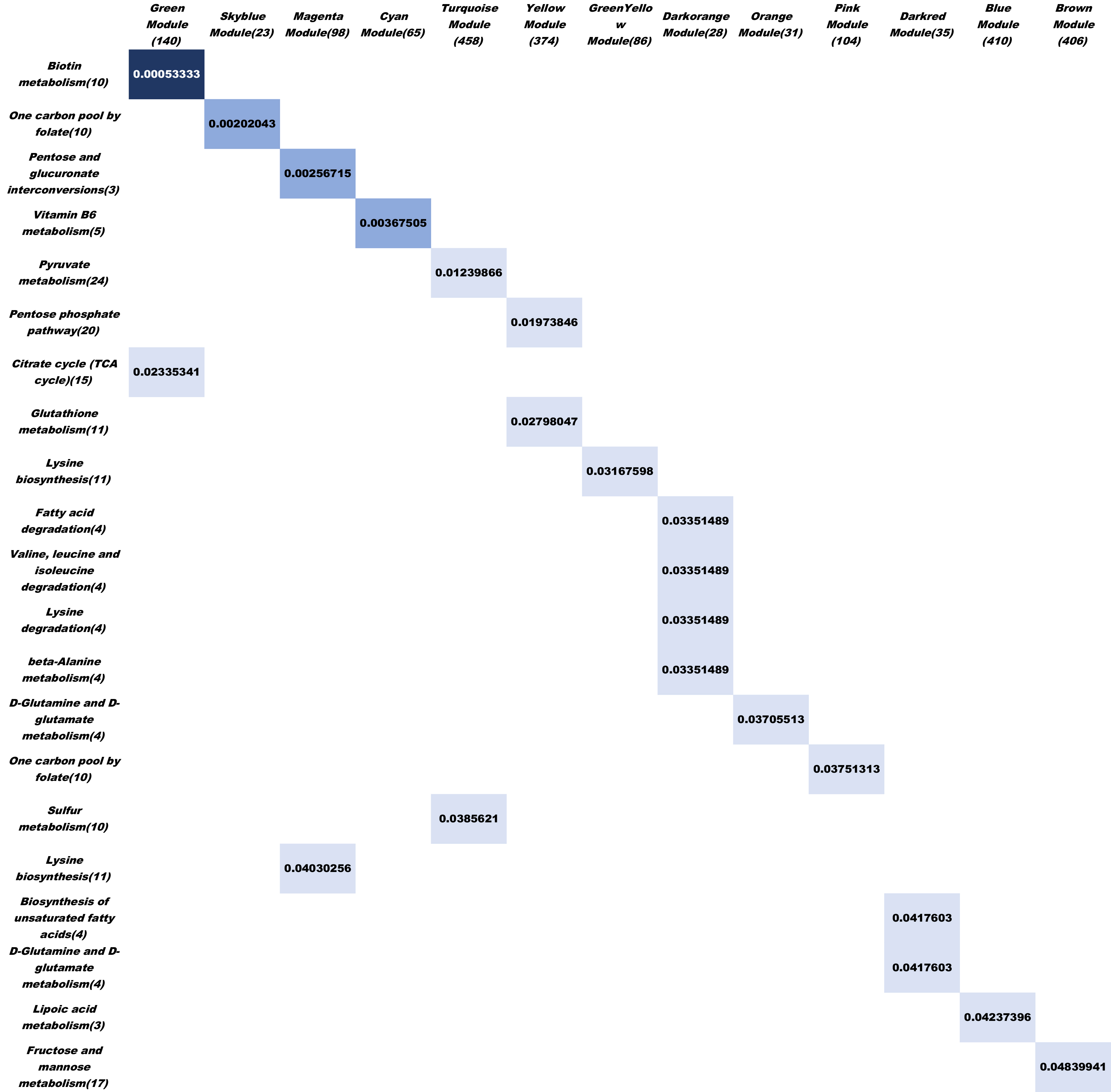
Gene coexpression module clustering
Partial correlation measure the direact interaction for each pair of genes. But in real genetic network, the secondary regulatory phenomen is really common. Although partial correlation matrix can measure “the direct distance” for each pair of genes, It will miss a lot of information when we need to describe the impact of one genes to the whole network.
Because of it, we decide to use hierarchical clustering method to cluster gene expression profile and get gene coexpression module. For clustering and clustering parameter optimization, see text2 for more details.
Each module represent one or some biological pathway
School's name:SCAU
Member's name:SCAU
Designed by:SCAU