Team:Toulouse-INSA-UPS/Model
MOLECULAR MODELLING

Why do we want to model our protein?
Our novel fusion protein contains three binding sites: Streptavidin (SAv), a Carbohydrate Binding Module (CBM3a) and an unnatural amino acid, azidophenylanine (AzF). These “heads” of our Cerberus are connected by flexible regions that are 30-50 amino acids each. The linkers constitute Intrinsically Disordered Regions of our protein, or IDRs, due to this flexibility. These are known complex domains that lack a fixed or ordered 3D structure, and that can occur when predicting and modelling large proteins.

If possible, defining a range within which the linkers remain would give us a better idea of how the protein behaves in situ. This task presents a particular challenge, as their structures have never been successfully resolved by crystallography.
As our linkers between domains are so flexible, it was possible that the CBM3a and SAv heads could be brought into contact. Their tertiary structures could also collapse due to new interactions between them. We needed to ensure that there will be no such interactions between these sites that would prevent them from binding to their intended ligands.
Incorporating an unnatural amino acid into a protein is a complex task not just for molecular biologists, but also for molecular modellers. Most protein prediction and modelling algorithms use predefined force field parameters for the 20 canonical amino acids, which simplify calculations. Using AzF meant building the 3D structure, minimising its potential energy then generating a specific force field for it. Rather than go through this whole pipeline multiple times, we opted to construct it bonded with the DBCO-Fluorescein, the fluorophore that we were using in the wetlab, from the start.
Finally, evaluating the potential interactions between ligands attached to our protein was the final step of our modelling process. For this, we had to evaluate the average distance between the different domains and its variations, which will also provide information about the maximum size of the ligands that we can use.
All of these questions can be answered through molecular modelling. However, each step requires its own approach and software. On this wiki page, we will detail these steps and our thought process behind the solutions that we chose to use.
How does (protein) molecular modelling work?
As biochemistry techniques give us a snapshot of the structure of a molecule, molecular modelling tends to integrate the multiple information and interactions possible within a molecule and between a molecule and its environment over time. In order to model a protein, we first need to understand the chemical basis forming it.
Protein structures roughly consist of a polyamide main chain with fuctional ramifications decorating it. The energetical stability of the bonds generally determines the underlying structure. It is the important to define the different source of conformational variability both within the main chain and on the lateral functions. Each of these many bonds present a large amount of variation, intoducing multiple dimensions in which the full molecule can evolve.

Figure 1: Peptide bond between two amino acids.
First, the distance between two atoms involved in a covalent bond can shift. Once three atoms are involved, we also need to study the angle separating the two distal atoms. Adding a fourth atom means needing to evaluate the torsion of the middle bond. This determines how far the fourth atoms deviates from the plane formed by the other three, on top of the previous parameters. This is considering the case where all four are connected sequentially. If not, the torsion is said to be improper.


Figure 2: Variables involved for neighbouring atoms (length of bond, angle, torsion, improper torsion)
These are the main factors to calculate for neighbouring atoms. When considering two distant atoms, we must also include a number interactions.
Hydrogen bonds are extremely common along the various folds of a protein, often maintaining its structure. If two atoms are close enough together, the Van Der Waals force plays a considerable role in defining the distance between them. Charged amino acids encountering another amino acid of opposite charge creates ionic interactions, which can appear or disappear depending on the pH of the surrounding environment.


Figure 3: Interactions between distant atoms (Van der Waals, ionic, hydrogen bonds)
When seeking the correct conformation of a protein, algorithms seek to minimise its potential energy. Minimisation algorithms therefore constitute a founding step through which all molecular modelling projects must go. A lot of different search methods exist to find local minima of the energy function of a protein. The most widespread use the first derivative of the energy function to find when it is equal to 0 (Steepest Descent, Conjugated Gradient). These are generally quite memory efficient for the computers running them, but have variable convergence speeds when approaching a minimum. A more complex solution would be to employ the Newton-Raphson method, which uses the second derivative of the energy function. It does however demand much more power from the computer, but is very accurate when close to the energy minimum.
Once a minimised structure of a protein is obtained, it is possible to run a molecular dynamics simulation to study it. This allows us to glimpse its behaviour over time. It is however a very long process, requiring many hours of calculations on a traditional computer. Upscaling it to multiple processors is generally compatible with molecular dynamics software, and very useful to save time.
By applying a starting velocity to each atom, even a weak one, we can observe how they will interact and move over time. This of course requires calculating values for the energy involved in each interaction listed above. Doing so requires vast amounts of calculations, and a few nanoseconds of simulation calculated requires hundreds of CPU hours. As proteins generally operate in a solution, representing the solvent and surrounding cofactors yields higher quality results at the cost of adding even more atoms to simulate.
It is important to remember however that this process will always remain a mathematical approximation of reality. As such, specific values like the exact strength of a ligand - receptor interaction or length of tertiary protein structures are near impossible to obtain, even in high precision simulations.
How did we model our protein?
3D structures of the CBM3a and SAv domains are available on the Protein Data Bank (PDB). We chose to use 4JO5 X and 4JNJ X, both obtained by X-ray diffraction, with a resolution of 1.98 and 1.90 A respectively.
After obtaining these files, we faced two problems. First, the linkers connecting our heads had never been resolved by crystallography, meaning that all sorts of interactions between the two could theoretically occur. Second, azidophenylalanine had never been modelled, in its native state or clicked to another molecule. This process will be detailed further on in this page.
We decided to start by modelling our Cerberus under the Orthos form, with a simple phenylalanine replacing the AzF residue. Orthos was Cerberus’ little brother in Greek mythology, with only two heads, like this version of our fusion protein. This task was completed through homology-driven prediction algorithms. The three we chose to use were I-TASSER, Swiss Model and MODELLER. This allowed us to obtain a 3D structure of our protein on which we could start calculations.
From their website: “I-TASSER is a hierarchical approach to protein structure and function prediction. It first identifies structural templates from the PDB by multiple threading approach LOMETS, with full-length atomic models constructed by iterative template fragment assembly simulations. Function insights of the target are then derived by threading the 3D models through protein function database BioLiP.”
From their website: “SWISS-MODEL is a fully automated protein structure homology-modelling server, accessible via the ExPASy web server, or from the program DeepView (Swiss Pdb-Viewer).”
From their website: “MODELLER is used for homology or comparative modeling of protein three-dimensional structures. The user provides an alignment of a sequence to be modeled with known related structures and MODELLER automatically calculates a model containing all non-hydrogen atoms.”
Only Modeller provided us with a satisfactory structure, most likely due to its improved de novo prediction capacities and the usage of restraints on the final model. The other two algorithms struggled to suitably resolve the position of the linkers.

Figure 4: Structure of our Orthos protein obtained with MODELLER.
The LAAS, Laboratoire d'Analyses et d'Architecture des Systèmes, a French National Scientific Research Centre, developed a novel software package called Psf-Amc (ref: A. Estana, N. Sibille, E. Delaforge, M. Vaisset, J. Cortes, P. Bernado. Realistic ensemble models of intrinsically disordered proteins using a structure-encoding coil database. Structure, in press.).
In robotics, by giving all the needed information about the various articulations in a mobile limb, AI software can calculate all positions which the arm can adopt. The LAAS applied this to proteins by defining subchains of the protein instead of individual atoms like other modelling software. By applying these possibilities and basic physics and chemistry equations to the different possible positions, they can very rapidly calculate hundreds of approximate models of the protein
Using the Orthos model, two algorithms were applied to it, the first using Single Residue Sampling (SRS) and the second using Triple Residue Sampling (TRS).







The figures shown directly above were generated by loading 5 different structures calculated by the LAAS, then aligning the CBM3a domain of each of them. This presents the stability of our protein.
As you seen on the figures, both of these simulations show the range of positions that the streptavidin head can take in relation to the CBM3a, also demonstrating the high flexibility of the linkers.
With these structures, we were able to choose the most probable overall conformation of our Cerberus protein to continue modelling. The next step was to add in the functional molecules that we wished to attach to our protein.
For the next step, we needed to determine a viable 3D model of azidophenylalanine. We opted to build it clicked onto a DBCO-fluorescein molecule, to simulate it functionalised from the start. This proved harder to accomplish than originally planned.
The basic chemical structure was drawn using Avogadro, an advanced molecular editor and visualiser, from a drawing of the AzF-DBCO-fluorescein molecule. It was also briefly minimised here for optimal geometric conformation. Next, LeAP, part of the AMBER suite (described below), was used to generate the force field of the molecule. This process calculates the various physical properties (mass, charge, energy, …) of each atom in the structure, to be reused for the molecular dynamics step.
The complex amino acid then had to be integrated as a part of the protein, which occurs quite rarely in protein modelling. Usually, canonical amino acids are used, which the modelling algorithms understand simply. However, here, we needed to align the structure of our AzF residue with that of the phenylalanine at the end of the C-Ter linker, remove the phenyl then add the AzF with all the correct bonds (and none of the wrong ones) to the neighbouring amino acids in the chain.
Biotin is a well known case study of ligand - receptor, so building and parametrising it was quick to perform. Again, we used the AMBER force field generating software to create the necessary files. Placing it into the correct position in the streptavidin binding site was used by aligning our streptavidin domain and aligning that with the original PDB file, which contained the biotin position obtained by X-ray analysis.
From their website: “The term "Amber" refers to two things. First, it is a set of molecular mechanical force fields for the simulation of biomolecules (these force fields are in the public domain, and are used in a variety of simulation programs). Second, it is a package of molecular simulation programs which includes source code and demos.”
From their website: “NAMD, recipient of a 2002 Gordon Bell Award and a 2012 Sidney Fernbach Award, is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on Charm++ parallel objects, NAMD scales to hundreds of cores for typical simulations and beyond 500,000 cores for the largest simulations. NAMD uses the popular molecular graphics program VMD for simulation setup and trajectory analysis, but is also file-compatible with AMBER, CHARMM, and X-PLOR.”.
The minimisation phase employs a different calculation method to determine the optimal local minimum with greater precision than previously. It consists of multiple sub-phases, with the first one holding the protein firmly to avoid deviations, then slowly releasing it to allow it to relax into a suitable position.
The heating phase brings the protein past the bumps in front of local minima to find a more global minimum. This is necessary for understanding how it reacts to temperature with energy available around and inside it.
The equilibration phase decreases the temperature of the protein and its solvent to allow it to settle back down into a local medium, usually a lower one than the previous minimum attained.
The production phase is when the main part of the simulation occurs. While the other phases last fraction of a nanosecond, this one takes place over tens of nanoseconds, sometimes up to a few milliseconds. This allows us to view how the protein structure changes over time with all the factors taken into account.
Simulations were conducted first on Amber18 for the minimisation, heating and equilibration phases of the dynamics workflow, and NAMD for the produciton phase due to its higher multiprocessor capacities. The first three steps were run on 80 processors parellelly, whereas production was performed on 1000.
Two separate production phases were performed. The first simulated the native protein. The second featured a constraint keeping the sulfur atom of the biotin molecule 7 A from the alpha carbon of the nearby XXX amino acid, to be sure to keep it within range of its associated binding site.

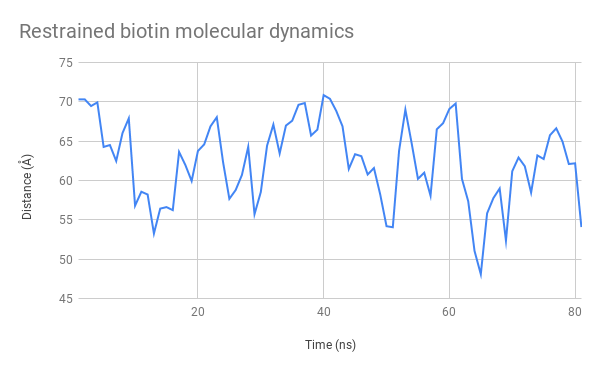
Above, you cans see a video generated from the structural data generated by our simulation. By observing its general structure, we can see that the linker sections are indeed very flexible but seem to adopt some preferred positions. For example, the N-terminal linkers sticks to one side of the CBM3a domain due to hydrogen interactions. We can also see the biotin molecule exiting its binding site after around 15ns.
We also tracked the distance between the alpha carbon of the azidophenylalanine residue and the XXX residue, in the binding site of the streptavidin domain. Extracting the raw numbers for each frame displayed allowed us to study its evolution closely, as you can see on these plots:

The Root Mean Square Deviation (RMSD) calculates the sum of the distances between each atom of a molecule, compared to the first frame of the simulation. Through this, we can study the varibility of our protein and, by selecting only specific residues, the variability of certain regions of it. This way, we can compare the data obtained for the structured domains and the IDRs to remark on the formers stability and the latters flexibility.

Conclusion
Calculations took a total of 100,000 hours of CPU time. Over 85GB of data was generated.
Thanks to these results, we are able to certify that our functional domains remain appropriately structured and retain their binding properties. Although we observe that the biotin molecule becomes released from the Streptavidin docking site, this is most likely due to small changes that occur in the arrangement of the various tertiary structures that compose it. However, it is important to remember that all of these data are obtained through mathematical approximations, and that it is important to verify them in situ. It would be extremely difficult to certifiably determine how affine for biotin our SAv domain is in the final structure through molecular modelling alone.
By measuring their RMSDs and following their positions over the duration of our simulations, we can confirm that our linker regions are too flexible to resolve a definite conformation. Despite this exteme variability, we never observed entanglement between them, nor did we see them bring the binding sites of our domains into contact. With this information, we can also be sure that the linkers shouldn't adopt a conformation where these binding sites become unavailable to their ligands.
Finally, tracking the distance between the two distal heads showed that we could indeed attach medium to large funcitonal molecules to these heads without risking too many unwanted interactions. However, due to the fact that they remain within 100 A of each other, we could still imagine placing functions on the heads designed to interact with each other, for example to subunits of a protein that meet and activate under certain conditions.
References
- E. Morag et al., "Structure of a family 3a carbohydrate-binding module from the cellulosomal scaffoldin CipA of Clostridium thermocellum with flanking linkers: implications for cellulosome structure.", Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013 Jul
- D. Demonte et al. , "Structure-based engineering of streptavidin monomer with a reduced biotin dissociation rate.", Proteins. 2013 Sep
No dogs were harmed over the course of this iGEM project.
The whole Toulouse INSA-UPS team wants to thank our sponsors, especially:









And many more. For futher information about our sponsors, please consult our Sponsors page.
The content provided on this website is the fruit of the work of the Toulouse INSA-UPS iGEM Team. As a deliverable for the iGEM Competition, it falls under the Creative Commons Attribution 4.0. Thus, all content on this wiki is available under the Creative Commons Attribution 4.0 license (or any later version). For futher information, please consult the official website of Creative Commons.
This website was designed with Bootstrap (4.1.3). Bootstrap is a front-end library of component for html, css and javascript. It relies on both Popper and jQuery. For further information, please consult the official website of Bootstrap.