Team:NCKU Tainan/Design
Design
Overview of Designed Pathway

Calvin-Benson cycle is one of the most important pathways for inorganic carbon to be converted into organic carbon in the carbon cycle. Plant, algae, and cyanobacteria utilize light as energy source for Calvin-Benson cycle. Taking the advantage of the pentose phosphate pathway, a native metabolic pathway of E. coli, only two additional enzymes will be needed to reconstruct the pathway in E. coli -- PRK and RuBisCO, which we will describe more in detail. The primary product of the pathway is pyruvate, which can be utilized to produce various valuable products.
Chassis Organism
We would like to test our pathway in various E. coli strains to see the functionality of our construction. We selected three different strains: BL21 (DE3), W3110, W3110 (L5T7). BL21 (DE3) is a common expression strain that is widely used to express recombinant proteins using T7 polymerase. We expected that the high production of protein may change the entire native metabolic pathway. W3110 (K-12 laboratory strain) is reported to be resilient in a stressed environment. We expected that W3110 will grow well even if the sole carbon source is xylose. W3110(L5T7) (provided by Dr. Ng) is a constructed lab strain based on W3110. T7 polymerase was inserted into its genome.
PRK
What is its function?
We first introduced phosphoribulokinase (PRK), an enzyme from cyanobacterial Calvin cycle, into the central carbon metabolic pathway of E. coli. PRK catalyzes the conversion of ribulose-5-phosphate (Ru5P) from the pentose phosphate pathway of the central carbon metabolism to ribulose-1,5-biphosphate (RuBP). One ATP is required for this conversion. The accumulation of RuBP in E.coli would cause cell growth arrest because E. coli can not metabolize RuBP.

How do we construct this part?
Steps involved in expressing PRK in E. coli. We initially confirm the gene sequence of Synechococcus elongtus PRK from NCBI gene database. We then codon optimized the sequence so E. coli can express the protein properly. The optimized sequence was sent to IDT for gene synthesis. We PCR amplified the gene fragments and digest it with restriction enzymes HindIII and SpeI. After digestion, we ligate the fragments into pSB3K3 plasmid with PLacI-rbs(B0034) located upstream of the fragment. The plasmid was then transformed into DH5 alpha.

How do we test its function?
We initially decided to measure the concentration of RuBP by HPLC. Our instructors pointed out some difficulties in HPLC measurement such as excessive noise signal in our sample. We then designed an experiment to prove the function of PRK in an indirect manner: by measuring its growth rate. RuBP, the product of PRK, is toxic to E. coli. We expressed this protein independently in xylose M9 to check cell growth. If the cell growth is arrested, we can indirectly conclude the function of PRK.
Rubisco
What is its function?
Ribulose-1,5-biphosphate carboxylase/oxygenase is one of the world’s most abundant enzyme. It catalyzes the conversion of inorganic carbon into organic carbon. In our designed pathway, the function of the RuBisCO is to convert Ribulose-1,5-biphosphate (RuBP) from the upper pathway and carbon dioxide into 3-phosphoglycerate (3PGA). 3PGA will then be converted to pyruvate by the native metabolic system of E. coli. After mining information from various publications, we selected RuBisCO from Synechococcus elongatus PCC7002, which is a well-studied cyanobacteria. Its genome is completely sequenced and it is often used as a model organism for gene manipulation. Previous research has utilized E. coli as a host of random mutagenesis to enhance the activity of Synechococcus RuBisCO.

How do we construct this part?
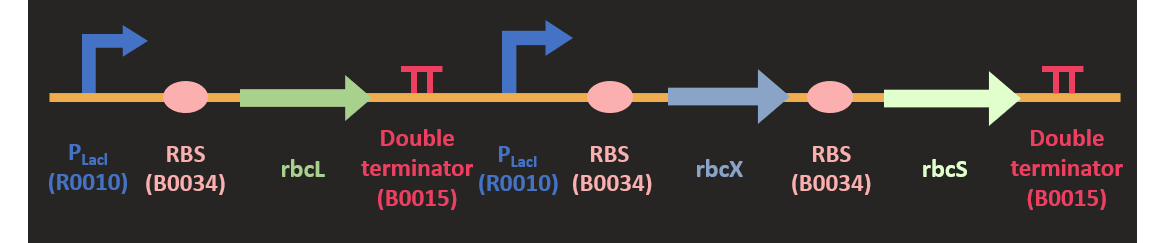
Akin to the construction of PRK, we codon optimized the sequence of three rubisco subunit and clone it into pSB1C3 plasmid with HindIII and SpeI. The sequence and the size of RbcL is much larger than other subunit, so we separated rbcL from rbcX and rbcS subunits. RbcX and rbcS is separated by a rbs (B0034) for the convenience of construction. We attached two different promoters upstream of the rubisco. They are PLacI and PT7 promoter. Since we would like to increase the expression of this protein in the metabolic pathway, we would like to test various promoter combination to find out the most efficient combination for our pathway.
How do we test its function?
Measurement of 3PGA or pyruvate concentration could not directly reflect the activity of rubisco since both of them are important metabolites that will flow to downstream metabolic pathway. We then decided to determine its function by total solution test which we will mention below.
CA
What is its function?
RuBisCO is the rate-limiting enzyme in carbon fixation. Oxygen competes with CO2 as a substrate for Rubisco, giving rise to photorespiration. To overcome this problem, some photosynthetic organisms have evolved their own carbon concentrating mechanisms (CCM), which helps to maintain a sufficient amount of CO2 around RuBisCO.
We are inspired by the carbon concentrating mechanisms (CCM) of cyanobacteria. In cyanobacteria, Rubisco and carbonic anhydrase (CA) is encapsulated in a microcompartment, the carboxysome. Carbonic anhydrase, also known as carbonate dehydratase, is involved in the interconversion between CO2 and HCO3-. This enzyme can be found in most organisms, including E. coli but the difference is its catalyzing rate in hydration and dehydration of CO2. Therefore, we will incorporate into our system the carbonic anhydrase gene from Synechococcus elongatus PCC7002.

How do we construct this part?
We first codon optimized the sequence and insert it into the empty pSB1C3 plasmid with HindIII and SpeI just as mentioned above. In our optimized sequence, we have already designed a PT7 promoter in front of CA, so we can directly ligate it into the plasmid. The constructed basic part is then linked with other basic parts to complete our construction.

How do we test its function?
To measure the enzyme activity of CA, we compare the conversion rate of carbon dioxide to bicarbonate ion. After saturated CO2 solution is prepared, we add fixed amount of bacteria broth that contains CA construction into the solution. We then measure the time interval of the decrease pH value from 8.3 to 6.3. We compare the measured time interval with the time interval that enzyme was not added to determine the enzyme activity of CA.
Dual Plasmid System
We decided to construct the whole pathway with the dual plasmid system. Previously, every basic part was the backbone conserved in the backbone of pSB1C3. We then link the construction together and even change the backbone of some composite parts to pSB3K3 for lower protein expression.
Rubisco whole protein in pSB1C3
We link each basic parts with biobrick standard method. We link PT7-rbcL and PT7-rbcX-rbcS together. The former, the insert, was digested with EcoRI and SpeI and the later, the backbone, is digested with EcoRI and XbaI. We ligate the backbone with the insert to complete this composite part.
Prk gene into pSB3K3
PRK catalyzes the reaction of turning Ru5P into RuBP. Not native to the host, RuBP is, nonetheless, toxic to E. coli. We hope that expression of PRK to be lower in the host so we change the backbone of it into pSB3K3. We selected J04450 from the distributed kit that under the backbone of pSB3K3, which will express red color after the formation of the colony. We digest both backbone and insert with EcoRI and PstI and ligate both fragments. We can then select the colony that does not present red color to prove that the ligation was conducted successfully.
Link Prk with CA into pSB3K3
We also constructed the composite part that contains both CA and PRK. We construct it using the method mentioned in rubisco whole construction. We cloned the fragments into pSB3K3 for lower expression of PRK.
Transformation
After the construction of various composite parts, we co-transform them into three E. coli strains: BL21(DE3), W3110, and W3110(L5T7). Since BL21(DE3) and W3110(L5T7) contains T7 polymerase, we co-transformed composite parts that contains T7 promoter into these strains. We co-transform plasmid that only contains LacI promoter into W3110.
How to prove our design?
We designed a total solution test to verify the function of our whole construction. We incubate the constructed strains in modified M9 medium that contains 0.4% xylose as its sole carbon source. The construction is designed to consume xylose as energy source and as a material for Calvin-Bensson cycle. We then measure the optical intensity (O.D. 600) to characterize the cell growth. At a fixed time interval, we use DNS assay to measure the sugar consumption of the bacteria. By comparing the experimental group to the control group, we can prove that our engineered strain utilize carbon dioxide as its carbon source.
pH sensing system
The pH sensing system, our side project, is a system that allows us to monitor the pH in the surrounding medium in our device at any time by observing the color change of the medium.
We selected two pH sensitive promoter from E. coli: Pasr and PgadA. PgadA will be induced under neutral condition while Pasr will be induced under acidic condition. We cloned a GFP and sfGFP gene downstream of these promoters respectively, whose product will express green fluorescence once the promoter has been activated. For the design of PgadA sensing system, we took the previous constructed biobrick from 2016 iGEM Dundee team, BBa_K1962013, as our reference. We also improve the PgadA biobrick to enhance the expression of GFP.
In conclusion, when the color of the medium turns from turbid yellow to green, it indicates the pH of the medium has altered so we can determine the pH condition of ethe medium.

How do we construct this part?
We first extracted whole genome DNA from E. coli MG1655 and amplify both promoters by PCR using primers that contains HindIII and SpeI. We then exchanged the promoter with the previously constructed plasmid that contains PT7 and GFP or sfGFP. We initially transformed the constructed plasmid into DH5 alpha for colony screening. We then transformed the plasmid into BL21(DE3) to test its function. We also design another biobrick that contains riboJ (a signal amplify fragment) at the downstream of PgadA to get the signal more clearly and enhance the specificity.

How do we determine its function?
We measure the fluorescence intensity of the plasmid in different pH environment to determine its promoter activity. We incubate the bacteria in pH modified M9 medium and measure the fluorescence intensity (absorbance: 480 nm, excitation: 510 nm).
References
- Z. Cai, G. Liu, J. Zhang, Protein Cell (2014) 5: 552.
- F. Gong, “Quantitative Analysis of an Engineered CO2 -Fixing Escherichia Coli Reveals Great Potential of Heterotrophic CO2 Fixation.” Biotechnology for Biofuels, BioMed Central, 18 June 2015
- “The Coupling of Glycolysis and the Rubisco-Based Pathway through the Non-Oxidative Pentose Phosphate Pathway to Achieve Low Carbon Dioxide Emission Fermentation.” NeuroImage, Academic Press, 25 Mar. 2015
- “Sugar Synthesis from CO2 in Escherichia Coli.” NeuroImage, Academic Press, 23 June 2016
- H. Cheng, E. J. Yang, Y. L. Liu, F. Y.Chenm, Y. Ou, S. Y. Li. “The Comprehensive Profile of Fermentation Products during in Situ CO2 Recycling by Rubisco-Based Engineered Escherichia Coli.” Microbial Cell Factories, BioMed Central, 2 Aug. 2016