Team:Stanford/Project
<!DOCTYPE html>
In 1997, Dove, Joung, and Hochschild showed that they could initiate prokaryotic transcription through the interactions of two proteins fused to the alpha subunit of RNA polymerase and a lambda repressor, respectively. The system fused the lambda repressor to a bait protein and the alpha subunit of RNA polymerase to a target protein (Figure 1). The interaction of the bait and target proteins stabilizes the binding of the RNA polymerase allowing the initiation of transcription [1]. This P2H (Prokaryotic two-hybrid system) developed by Dove et al was used primarily as a way to screen protein-protein interactions, and was spun into the BacterioMatch kit produced by Agilent [2]. Its utility as a detection mechanism, however, remained unexplored.

Inspired by the yeast two-hybrid system designed iGEM Tsinghua 2017 [3], we fused antibodies and dCas9 proteins to the subunits of the P2H system, creating a versatile platform capable of detecting specific DNA sequences, small molecules, and proteins. Our DNA detection system works with dCas9-protein fusions, which can be targeted to specific DNA sequences (Figure 2), and is called casP2H (cas9 prokaryotic two hybrid system). Our protein and small molecule system works by fusing single-chain-antibodies specific to different epitopes of a target protein or small molecule, and is called SCAP2H (single chain antibody prokaryotic two hybrid system).
This detection system is unique because it utilizes transcription. Detection-induced transcription allows genes to be activated by the presence of a user-selected molecule, therefore creating a genetic output to a chosen input. For example, a therapeutic protein could be transcribed in response to a disease biomarker.
For a narrative of how we arrived at this project, see our Integrated Human Practices page.
The CRISPR/Cas9 system is a new tool that has tremendous power for the future of bioengineering and synthetic biology. A number of projects have successfully harnessed this technology in applications spanning both gene expression manipulation and DNA detection. While historically DNA detection has been carried out via polymerase chain reaction (PCR), the specificity and adaptability of the Cas9 protein to selectively target DNA has opened the window to a new and streamlined strategy for detecting DNA fragments in vivo.

Deactivated Cas9, or dCas9, is an adapted form of the Cas9 protein that is designed to sit on the DNA molecule without initiating a cut in the double stranded base pairs as the active Cas9 protein normally does [4]. The 2015 Peking University iGEM team designed a split luciferase dual-dCas9 system that effectively targets fragments of DNA [5]. In their system, two dCas9s simultaneously bind different neighboring sections of the target DNA, and in doing so bring together two halves of a luciferase molecule for a visible indicator of DNA presence. This project was successfully executed and optimized to reliably and adaptably detect DNA fragments of varying lengths.
Our project builds on this dual-dCas9 system by implementing a transcriptional-based reporter system by bridging dCas9 with the prokaryotic two-hybrid (P2H) system. Instead of split luciferase as an indicator, in our system the proximity of dCas9 binding brings together a larger protein complex (adapted from the P2H system) and initiates transcription of red fluorescent protein (RFP) (Figure 2).
While this initial design accomplishes a similarly goal to the 2015 Peking project of creating a fluorescent indicator for the presence of DNA, the key difference is in the adaptability of transcriptional activation as a reporter—instead of luminescence as the sole output of the system, our system can be expanded beyond detection by exchanging the RFP output for other relevant proteins for applications (see applications section below) in targeted therapeutics, bacterial strain engineering, and beyond.
We are adapting a prokaryotic two-hybrid system and combining it with dCas9 in order to selectively detect DNA sequences for disease diagnostics. In order to ensure specificity of sgRNA to dCas9 binding, we used two different dCas9 proteins derived from streptococcus pyogenes and streptococcus aureus bacteria. The pyogenes dCas9 was fused via a tri-alanine linker to a lambda repressor that binds the pOL-62 promoter region 35 base pairs upstream of the RFP initiation site. The aureus dCas9 was fused via a tri-alanine linker to the alpha-subunit of RNA polymerase, which in turn recruits polymerase for transcription. The two sgRNA sequences are produced separately and bind selectively to the dCas9 molecules and target DNA, thus bringing together the entire complex and activating RFP.

We are not pulling the idea directly from a single paper, but instead combining literature about the function of dCas9s, the prokaryotic two-hybrid system developed by Dr. Ann Hochschild at Harvard, and the previous successful research executed by Peking iGEM 2015 using a similar two hybrid dCas9 system to join a split luciferase. Additionally, after finalizing the concept we sat down with Dr. Stanley Qi, a CRISPR scientist at Stanford, to confirm idea feasibility and get guidance on how to best execute the sgRNA design.
Although in our original design both fusion proteins utilized pyogenes dCas9s, Dr. Qi recommended switching one of them out for aureus dCas9. Without this change, even though we would produce two different sgRNA strands that had distinct 20 bp variable regions (highlighted in orange and red) to bind the target DNA, due to random chance 50% of the time both pyogenes would be bound to the same type of sgRNA and therefore not be able to simultaneously bind the target DNA (Figure 3).

Using scientific research, iterative design, and expert consultation, we arrived at the Cerberus Detection for DNA system via the biodesign process (see Human Practices for more info).
The DNA detection system requires two fusion proteins, sgRNA, a modified RFP reporter, and target DNA. Due to the origin of replication limits described in the SynORI 2017 iGEM project last year, it is necessary to limit bacterial transformations to only three plasmids. In order to deliver five DNA constructs with only three plasmids, we used Gibson Assembly to clone our constructs onto the shared plasmids (Figure 4).

While the original idea was to heat shock in linear fragments of target DNA to detect, after consulting with graduate students familiar with bacterial transformations we determined that this strategy would potentially be ineffective due to bacterial recombination altering the target DNA sequences. Therefore we instead chose to target a whole GFP plasmid that we could guarantee would be transformed into the cell without sequence alteration.
We specifically chose to target a GFP plasmid because we could then confirm binding of each dCas9 protein independently. For example, adding just the plasmid with the adCas9 fusion protein plus the sgRNA (far right in Figure 4) and the GFP plasmid leads to knockdown of GFP but no RFP transcription, thus proving successful targeting and binding of the sgRNA and pyogenes dCas9.



When initially discussing the system with Dr. Qi, we asked the question: What is the ideal spacing length between the two regions we are targeting on the GFP plasmid? While Dr. Qi could recommend having a minimum spacing region of about 20 bp given the size of the dCas9s, the actual ideal spacing distance could be anywhere from 20 to 100 base pairs and was a statistic that needed to be optimized for our specific system.
Additionally, there were three possible orientations in which the two dCas9 molecules could land on the molecule depending on whether they bound to the coding or template strand of DNA. Given the orientation and inter-sgRNA-target spacing length, we devised six different sets of sgRNA constructs to test each dCas9 orientation at a short (20 to 40 bp) and a long (50 to 90 bp) interspace region (Figure 5)
One limitation of the CRISPR/Cas9 system is that a protospacer adjacent motif (PAM) sequence must be present in the target DNA directly upstream of the 20 bp targeted region. While pyogenes dCas9 has a short and relatively frequent PAM of NGG (N = any nucleotide), aureus dCas9 has a PAM of NNGRRT (R = A or G) that is more specific and therefore less frequent.
When presented with a finite target DNA sequence, it can be tricky to find binding sites for aureus and pyogenes dCas9s that are in the desired orientation and have roughly the desired interspace binding region. To expedite this process, our team designed a computer software in which you can input the desired interspace region range, the desired orientation, and the specific PAM sequences for the dCas9 derivatives that are being used. The software then returns a set of all possible binding site combinations.
It is important to note that given the two 20 base pair target regions, plus the two PAM sequences necessary in the target sequence, there is an absolute minimum target DNA strand length of roughly 50 base pairs. At this length, however, the probability of finding the two PAMs in a desired orientation is slim. We designed a model to determine how many base pairs on average are required to have a 95% chance of the fragment being targetable. Our model can be found here
.Our team received aureus and pyogenes sgRNA constructs from Dr. Marie La Russa, a postdoctoral researcher in Dr. Qi’s lab. After modifying the variable 20 base pair regions in these sgRNA sequences on Benchling in order to target the GFP plasmid, we added bacterial promoters and terminators for both constructs and then ordered the DNA fragments from IDT.

The terminators L3S1P21 and L3S1P22 are derivatives of the Trrnb and were obtained from Chelsea Hu’s recent research on bacterial terminators [7]. Promoters J23100 and J23119 were taken from the Anderson promoter collection on the iGEM Registry [8].
Cerberus DNA Detection’s strength lies in its versatility. Via a simplified PCR with Agilent’s QuikChange Site-Directed Mutagenesis procedure [9], the variable regions in the sgRNA sequences can be easily swapped out to bind the dCas9s to any targetable DNA fragment. Although our initial design of targeting GFP prioritizes proof-of-concept over application, our team collaborated with synthetic biologists and people in fields outside of biology throughout our design and development process to make sure our product was a tool that would actually be used. We focused on developing three specific applications of this tool, but this should not box in its future uses.
Firstly, our system enables local delivery of therapeutics to specific infected areas. Our project inspiration was originally derived from this need area. We talked to local cannabis growers in the Bay Area and found out that crop loss to aspergillus flavus has significant impacts on both small and large scale growers (see Human Practices for more information). With Cerberus, therapeutic RNAi can be delivered to fight aspergillus flavus with specificity. RNA interference (RNAi) is a biological process in which a strand of RNA binds to its complement, creating a strand of double-stranded RNA. Cellular enzymes then cleave this double-stranded complex, leading to gene silencing. This process has been exploited by bioengineers in applications such as pest control to silence the pest’s vital genes and kill it. RNAi based pest control requires no laborious genetic engineering of crops—for instance, Syngenta’s RNAi based insecticide is a spray that contains double-stranded RNA. However, such a spray would need to be reapplied and can be costly to produce. With our system, we can enable screening of large amounts of crops for Aspergillus flavus by screening for DNA fragments commonly exchanged in bacterial recombination between e. Coli and the fungus. We could thus selectively deliver an RNAi-based fungicide to infected plants, rather than the entire population to save time and money.
Secondly, diseases such as liver cancer have biomarkers that can be exploited to improve diagnostic tools. Using our detection system will allow us to create a transcriptional response to oncogenic biomarkers. We spoke to Ritish Patnaik about his work identifying liver cancer biomarkers in the Wang group at Stanford University, and discussed how our system might be helpful by creating transcriptional data for his magnetoresistive (GMR) platform. This platform for early detection binds to transcribed RNA and intensifies the signal with magnetic amplification, which could lead to earlier and more sensitive liver cancer diagnoses.
Lastly, we had a conference call with Mehmet Berkmen, a senior scientist at New England Biosciences (NEB), to discuss potential applications within the field of large scale protein production. At NEB, batches of protein are synthesized by E. coli that mass produce proteins. Although antibiotics are used to select for bacteria that have the plasmid containing the desired protein, due to bacterial recombination it is common for strains to arise that lose the protein sequence without losing antibiotic resistance. Our system could be used to protect against this loss by using Cerberus to target the specific protein DNA and initiate transcription of resistance to an additional selection agent.
For our protein test case we chose to detect human IL1B, a protein expressed in response to illness and fever. We selected IL1B because it is an important human immunoprotein with multiple well-characterized antibodies. We fused two single chain antibodies targeting different IL1B epitopes (BBa_K2876015 and BBa_K2876011) to the lambda repressor and alpha subunit of RNA-polymerase III (BBa_BBa_K2876002 )described in Dove, Joung, and Hochschild in order to create two fusion proteins capable of activating transcription in the presence of IL1B (Figure 7). For our reporter we used the previously described pOL2-62 from Dove, Joung, and Hochschild fused to a RFP (BBa_K2876000). See more information about our reporter below.
We validated our IL1B with an ELISAwhich proved production of IL1B, and that the IL1B produced was folded properly so that antibodies could bind. Validation of antibody binding was essential to ensure that IL1B could function as the target protein in our Single-Chain Antibody Prokaryotic Two Hybrid Detection System (Figure 7).


Protein was extracted using a B-PER protocol. We used the negative control to calculate a null hypothesis for the log of absorbance when there is no protein. H0 Absorbance = Log(0.082) = -1.086. We then did a right-tailed hypothesis test to see if our values of Human Interleukin-1 Beta were statistically significant. The z-score was 21.57; the cutoff for statistical significance was a z-score of 1.895. Therefore, our data was statistically significant and we can accept the alternative hypothesis, Ha: Absorbance ≠ -1.806. This leads us to conclude that IL1B protein recognizable by IL1B antibodies is being made.


For our small molecule test case we chose to detect aflatoxin, a small molecule produced by Aspergillus fungi species that infects many agricultural crops, including cannabis.2,3 Aflatoxin also causes a number of health problems in people when inhaled or ingested by immunocompromised patients. We fused two single chain antibodies targeting aflatoxin (BBa_K2876005 and BBa_K2876003) to the lambda repressor and alpha subunit of RNA-polymerase III (BBa_K2876002) described in Dove, Joung, and Hochschild in order to create two fusion proteins capable of activating transcription in the presence of aflatoxin (Figure 11).

For our reporter we used the previously described pOL2-62 from Dove, Joung, and Hochschild fused to a RFP. See more information about our reporter below.
For their reporter system, Dove, Joung, and Hochschild used a strain of E. coli susceptible to knockout of histidine production in the presence of 3-AT [1]. Only when a protein-protein interaction initiates transcription of an alternate histidine production pathway would E. coli be able to grow on his-knockout 3-AT media [1]. Because the reporter in the Dove et al system only gave a binary response of (growth if interaction) or (no growth if no interaction) we felt it was not sensitive enough for our purposes. We contacted Ann Hochschild, who was kind enough to send us the sequence for the promoter for their system, pOL2-62 ( BBa_K2876000 ), a pLacZ promoter with a modified upstream region for lambda cl binding. We then took that promoter and linked it to a turbo-RFP, to allow for a concentration dependent fluorescence response (Figure 12). To test the reporter we transformed DH5-alpha with the pOL2-62 + RFP reporter plasmid, as well as the positive control bacteriomatch lambda and alpha plasmids from Addgene and induced with IPTG. Unfortunately, when we ran the experiment on the plate reader we saw no fluorescence response (Figure 13).




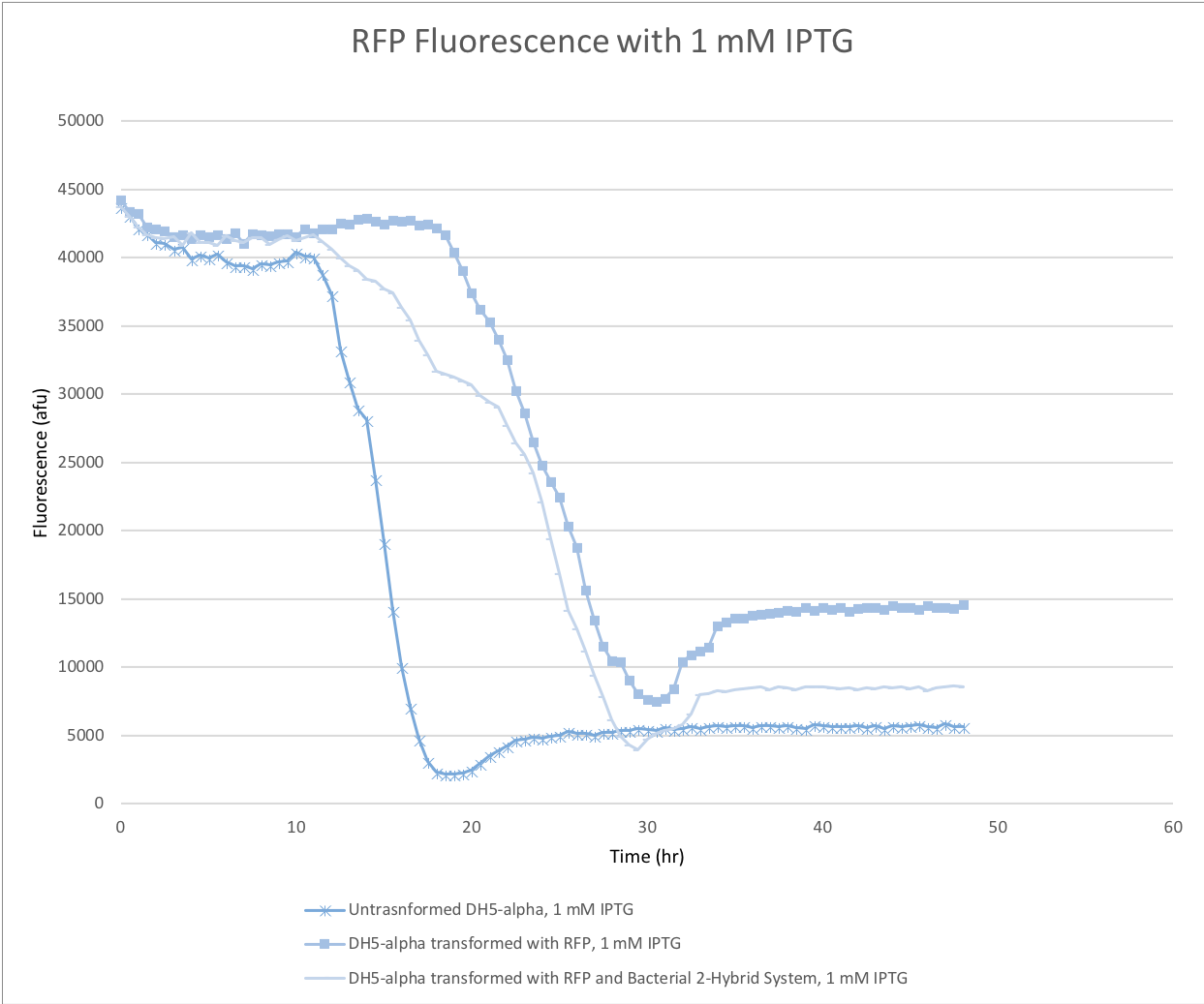
 Fig 13 - RFP-reporter fluorescence over 48 hours
Fig 13 - RFP-reporter fluorescence over 48 hours
1. Dove SL, Joung JK, Hochschild A. “Activation of prokaryotic transcription through arbitrary protein-protein contacts.” Nature. 1997;386(6625):627-30.
2. “BacterioMatch II Two-Hybrid System Vector Kit.” Agilent Technologies Instruction Manual.
3. https://2017.igem.org/Team:Tsinghua
4. Dominguez A A, Lim W A, Qi S L. Beyond editing: repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nature Reviews; Molecular Cell Biology. 2016; 17:5–15.
5. https://2015.igem.org/Team:Peking/Design/PC_Reporter
6. Ran F. Ann , Cong L, Yan W X. “In vivo genome editing using Staphylococcus aureus Cas9.” Nature. 201;520:186–191.
7. Hu, Chelsea Y et al. “Engineering a Functional Small RNA Negative Autoregulation Network with Model-Guided Design.” ACS Synth. Biol. 2018;7,1507−1518.
8. http://parts.igem.org/Promoters/Catalog/Anderson
9. “QuikChange II Site-Directed Mutagenesis Kit.” Agilent Technologies Instruction manual.