Team:UCSC/Design
Design
TARGET ORGANISM: Yarrowia lipolytica
Shortly after the choosing the project, searching for a potential host began. Initially, we held a meeting with Tom Sousa, a 1st year graduate student in the Bioinformatics Department at UCSC, and an expert on yeast. Tom recommended that we look into using Saccharomyces cerevisiae, aka brewer’s yeast. S. cerevisiae is special because it is extremely well studied and is ideal for biological research because of its short doubling time, ability to be transformed, and can be grown as a haploid.
In order to ensure that we were picking the best possible strain of yeast, a few team members continued research on different strains of yeast. Soon after, we discovered Yarrowia lipolytica, another “Generally Regarded as Safe” (GRAS) organism [1,2]. Although less studied than S. cerevisiae, Y. lipolytica has several benefits that led us to believe that it would be a better choice for our project:
- Y. lipolytica outperformed all alternative hosts in heterologous protein expression and high transformation frequency during homologous recombination of linearized plasmids [3].
- Y. lipolytica can survive on unconventional carbon sources like milk, glycerol, and oil [4,5].
- Y. lipolytica exhibits low side-product formation and high substrate specificity, which is ideal for producing a single product like progesterone[6].
Yeast that use the DNA repair mechanism homologous recombination (HR) are commonly chosen for engineering purposes because HR is efficient for inserting genes. Y. lipolytica has a propensity for a less favorable DNA repair mechanism, non-homologous end joining (NHEJ). To circumvent this problem, we searched for a strain that had the NHEJ mechanism, which is is usable when the KU70 gene is knocked out.
Our project required auxotrophic markers for selective screening of our transformants, so we looked for a ura3 and leu2 auxotroph. A paper by Verbeke et. al in 2013 described the JMY2394 strain, which satisfied our parameters, but was unavailable to us. We searched for a similar strain and settled on FKP393 by recommendation of Erin Bredeweg of the Pacific Northwest National Laboratory. FKP393 is a NHEJ mutant derived by transformation of Po1g with the specifications matA; leu2-270; ura3; xpr2-332; axp-2; ku70::hph+. We received FKP393 as a gift from the Fungal Genetics Stock Center at the Kansas State University. This strain includes all of our specifications but lacks sufficient annotation. We were informed by Cory Schwartz of UC Riverside that this was not an issue because the parent strain is the well-studied W29, which we can use for genome analysis.
PROGESTERONE METABOLIC PATHWAY

Figure 1: Simplified version of the reference pathway for progesterone biosynthesis in mammals, from the Kyoto Encyclopedia of Genes and Genomes (KEGG)[10].
Due to our extensive research on world issues, we chose to create a contraceptive in yeast: progesterone. Progesterone is a hormone produced by all mammals (men and women) naturally, and aids in many functions regulating fertility, menstruation/ovulation, pregnancy, and normal body function. "Progesterone prepares the endometrium for the potential of pregnancy after ovulation. It triggers the lining to thicken to accept a fertilized egg. It also prohibits the muscle contractions in the uterus that would cause the body to reject an egg. While the body is producing high levels of progesterone, the body will not ovulate" [7]. Progestins are routinely used in contraceptive pills, vaginal rings, and IUDs. Progestins are a chemically formulated version of progesterone that acts on progesterone receptors to increase cervical mucus and suppress ovulation to prevent pregnancy [8]. They are not naturally made by the human body and therefore have additional side effects on the women who take them [9]. Progesterone is a molecule naturally made by the body which can perform the same function as progestins: preventing pregnancy. Progesterone can do so without the harmful effects that progestins cause [9]. In mammals, the progesterone biosynthesis pathway begins with cholesterol. Enzymes convert cholesterol to pregnenolone and then to progesterone. Figure 1 shows a highly simplified version of the reference pathway for steroid hormone biosynthesis in mammals from the Kyoto Encyclopedia of Genes and Genomes (KEGG) to highlight the progesterone biosynthesis pathway [10].
The organism we are engineering, Y. lipolytica, does not produce cholesterol naturally but does produce a cholesterol precursor: zymosterol. In Y. lipolytica however, only the zymosterol-ergosterol pathway exists. We considered engineering three non-native enzymes (circled in blue in Figure 2) into Y. lipolytica to complete the zymosterol-cholesterol pathway, due to the common pathway to progesterone in mammals starting from cholesterol. However, because ergosterol is the yeast’s preferred pathway product, navigating through the cholesterol pathway would require completely knocking out the pathway to ergosterol (starting from ERG6 on the right). The addition of those three genes, the additional genes needed to take cholesterol to progesterone, and a pathway knockout required too much engineering for the time frame of our project. The enzymes highlighted in green exist in Y. lipolytica, see the end product of ergosterol circled in pink. The enzymes highlighted with white do not exist, and would need to be engineered into the organism.

Figure 2: End of pathway leading to Steroid hormone biosynthesis pathway starting from Glycolysis, to Terpenoid backbone biosynthesis, to Steroid biosynthesis in Y. lipolytica. Green highlight shows that those enzymes exist naturally in Y. lipolytica and white highlight means KEGG does not have enough evidence to show they exist in Y. lipolytica.
We needed to figure out how to synthesize progesterone in a creative way. We found that we could establish a synthetic pathway to progesterone directly from ergosterol (circled in pink in Figure 2). This was beneficial because that pathway exists naturally in Y. lipolytica. In Y. lipolytica and other fungi, ergosterol serves similar functions in fungi as cholesterol does in animal cells [11], meaning that while the pathway does not currently exist in the organism, it is possible to convert ergosterol to progesterone. We searched through research articles and found the work of Duport et al. from 1998 in which they outlined the possible pathway to convert ergosterol to progesterone using five genes[12]. The first gene is for the enzyme ∆7-sterol reductase (∆7Red), isolated from Arabidopsis thaliana, which reduces ergosterol to ergosta-5-enol and ergosta-5,22-dienol [13]. The next gene encodes for bovine side-chain cleavage cytochrome (P450scc), which converts ergosta-5-enol and ergosta-5,22-dienol to pregnenolone. P450scc requires the assistance of bovine ferredoxin-1 (FDX1) as an electron carrier, and adrenodoxin reductase (ADR) to reduce FDX1. The final gene is type II human 3β-hydroxysteroid dehydrogenase (3β-HSD) that converts pregnenolone to progesterone [14,15]. See the complete pathway to progesterone biosynthesis in Y. lipolytica below in Figure 3.

Figure 3: Progesterone biosynthesis pathway optimized for use in yeast. Design idea came directly from Duport et al., and genes for enzymes and electron carriers were codon optimized for use in Y. lipolytica[12].
We found the sequences for each gene from either KEGG or UniProt and codon optimized them for Y. lipolytica using the DNA works system from the High-Performance Computing at the NIH website[10, 16].
PROMOTER & TERMINATOR
Once we had constructed the pathway we wanted to use, we needed to choose a promoter and terminator. A favorable promoter or terminator is short in length with high levels of expression. We designed the genes in the biosynthesis pathway to function monocistronically, where each gene possesses its own promoter and terminator.
Promoter
Initially, we considered a galactose-inducible promoter, GAL10, to control the expression of the progesterone gene cassette. However, the presence of glucose represses GAL10 via the MIG1 binding site, and we wanted our yeast strain to be able to produce progesterone when grown on a variety of substrates, including glucose. To use this promoter, we would have to modify the MIG1 binding site for the promoter to function on glucose-enriched media. According to Dr. Hans Ronne of the Department of Forest Mycology and Plant Pathology at the Swedish University of Agricultural Sciences, maintaining galactose inducibility would entail a risky manipulation of the CYC8-TUP1 co-repressor complex.
We researched other methods of eliminating glucose-mediated repression, but deemed it more practical to use a constitutive promoter like TEF1. Constitutive promoters enable constant gene expression, while inducible promoters turn gene expression on or off depending on a certain factor. Controlling the amount of product produced is a critical element in creating a specific dose of medicine. To control dosage the consumer could heat-shock the yeast after a time calculated from the progesterone biosynthesis rate, as opposed to controlling the amount of galactose in the growth media.
Terminator
We originally chose the Tsynth27 terminator recommended by Dr. Hal Alper1 for its short length (66 bp) and high gene expressivity [17]. When we ordered the sequence from Integrated DNA Technologies (IDT), we received an error regarding the length of the efficiency region; the long, repetitive sequence of TATA base pairs, which was originally designed for higher efficiency, made synthesis of the gene block impossible.
We decided to use another short synthetic terminator, Tsynth8, also designed by Dr. Alper. Tsynth8 contains a shorter efficiency region yet still allows for high gene expression and low cryptic promoter activity in Y. lipolytica.
GENE CONSTRUCT ASSEMBLY
Initially, we determined that Gibson Assembly (GA) was the best option for plasmid cloning for two reasons: our mentors and team members have prior experience with the method, and GA can join many DNA fragments of any length. However, we wanted to explore other possible methods of cloning before making our final decision. We reached out to Dr. Jean Marc Nicaud and he suggested we consider Golden Gate Assembly (GGA) for plasmid construction. Dr. Nicaud informed us that GGA was an ideal method for Y. lipolytica plasmid assembly. Dr. Rohinton Kamakaka also advised us to use GGA as it offers a seamless, single-reaction assembly of DNA fragments, similar to GA[18].
After further research, however, we discovered multiple issues with this assembly technique: our team members and mentors have no prior experience with GGA, the process takes up to six hours, and we would need to insert unique type II restriction sites between our gene blocks, which adds another step to our project [19]. For these reasons, we ruled out GGA as our cloning method and returned to GA. GA requires that DNA fragments have homologous ends (either naturally or artificially) for assembly to occur. These ends are called Gibson overhangs (GOs). To create homology where it does not occur naturally, we will use primers with GO flags on either end that match the overhangs and homology regions on corresponding fragments. GA shortens the construction process because it does not require the use of restriction sites, can be performed in under an hour, and seamlessly integrates fragments.
Dr. William Belden informed us of his extensive experience with another assembly method: yeast-mediated cloning (YMC). At the beginning of our research, we considered this to be an innovative cloning technique, but we abandoned it due to our lack of experience. Dr. Belden explained that YMC works similarly to GA, but the process occurs naturally within yeast. Similar to GA, YMC requires homologous overlaps between fragments. Because GA and YMC involve similar designs, we reasoned that we could perform both experiments simultaneously without sacrificing much time or effort. Running both experiments in parallel will allow us to experiment with YMC while keeping GA as a contingency plan. We consulted Dr. Kamakaka on our parallel experiment idea, and he offered his support by providing protocols for YMC.
YEAST TRANSFORMATION
We want to integrate our gene cassette into the Y. lipolytica genome so that our construct will be passed on to offspring with each round of replication [20]. We investigated a paper on using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 to insert pathway genes into Y. lipolytica[21] and considered using this system to incorporate our progesterone precursor genes into the yeast genome. In this paper, researchers used CRISPR/Cas9 to integrate a set of two genes into the Y. lipolytica genome, but they used a gene cassette much smaller than ours will be [21]. Schwartz et al. (2017) identified five loci within the Y. lipolytica genome with a high rate of integration via CRISPR/Cas9-mediated homologous recombination [22]. They found that gene disruption in these regions did not affect the growth rate of Y. lipolytica, which made us consider using them to integrate our genes of interest.
Gene integration via CRISPR could cause gene drive, which occurs when a particular set of genes spreads rapidly throughout the population of an organism [23]. The use of gene drive violates the iGEM competition rules, so we will not use it in this project. To avoid gene drive, our mentor, Dr. David Bernick, suggested that we use microinjection of the Cas9 protein. However, micropipettes cannot penetrate the cell wall of Y. lipolytica, which makes this technique unusable [24]. The authors of Riveline and Nurse (2009) offered an alternative method that employs microfabricated channels to hold individual yeast cells while a piezoelectric unit shears them [24]. With the limited timeframe of our project, this option seemed too complex, so we abandoned CRISPR/Cas9 as our method of gene integration.
We also considered using homologous recombination (HR) to integrate our genes of interest. HR requires that our gene cassette be flanked by 1 kilobase (kb) regions homologous with a location in the Y. lipolytica genome. When a double-strand break naturally occurs within the cell, the cell can repair the break using homologous pieces, known as homology arms (HAs). HAs neighboring our genes of interest will signal the cell to repair the break and integrate our genes into the genome. Although HR is a great option for genomic integration, Y. lipolytica performs NHEJ more often than HR. In a typical strain of Y. lipolytica, using HR dramatically increases the time needed for our genes of interest to incorporate into the genome. Fortunately, Verbeke et al. (2013) showed that HR rates in Y. lipolytica increased by 56% when they knocked out the KU70 gene[25]. The KU70 gene regulates the NHEJ pathway in Y. lipolytica, and eliminating it forces the yeast to repair breaks using HR. We chose a strain that has KU70 knocked out for this reason, so HR is a viable option for us.
Cre-lox site-specific recombination is another method we explored for genomic integration. This technique uses a recombination enzyme native to bacteriophage P1, Cre recombinase, and lox sites that the recombinase targets [26]. Typical Cre-lox recombination uses two identical loxP sites which may invert or delete the genomic information between them depending on lox site directionality [27]. We found a publication by Watson et al. (2008) where they used two incompatible lox sites for recombinase-mediated cassette exchange (RMCE) [26]. In their genome, two incompatible lox sites flanked a selectable marker. The same two lox sites flanked the gene of interest in the plasmid.
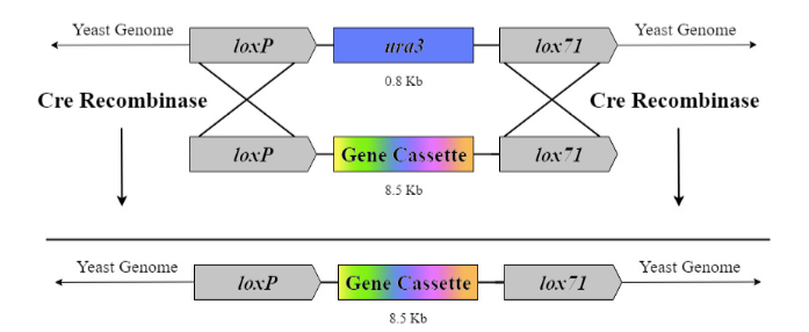
Figure 4: Cre-lox recombination method. Two incompatible lox sites, loxP and lox71, will flank selectable marker ura3 in our egineered strain of Y. lipolytica, and will flank the gene cassette. The regions between the lox sites will be exchanged when Cre recombinase is introduced.
Using two incompatible lox sites allows the Cre recombinase to recombine only the sequence between the lox sites; we determined that this system may work to incorporate our genes of interest. Figure 4 shows how RMCE works with two incompatible lox sites in the context of our experiment. We will use BioBrick loxP (BBa_J61046) and lox71. To confirm that Cre-lox has worked, we will engineer the selectable marker ura3 between the lox sites in the genome, and grow the transformed strain on uracil-enriched 5-FOA media to ensure only transformed colonies are viable.
Since we were uncertain of the size of the gene insert we could reasonably incorporate using RMCE, we contacted the authors of the article, Dr. Adam Watson and Dr. Tony Carr of the University of Sussex. They informed us that we could insert our genes of interest with little difficulty, as they previously used RMCE to insert up to 16 kb fragments with no decrease in efficiency.
After much consideration, we decided to use both Cre-lox recombination and HR in parallel experiments to determine which method yields the most recombinants. We will use HR to incorporate the lox sites into the Y. lipolytica genome, and HR to integrate the progesterone pathway. In a parallel experiment, we will use Cre-lox recombination and RMCE to insert the genes into the Y. lipolytica genome. We originally intended to characterize the recombination rate of the lox sites to satisfy one of the iGEM bronze medal requirements, while keeping the HR technique as a contingency plan if Cre-lox recombination fails.
QUANTIFICATION of PROGESTERONE
Several quantification methods were evaluated based on their specificity, accuracy and time efficiency. Each of these methods, additionally, require the purification of the products of the synthetic pathway from the yeast cells. We considered gas or liquid chromatography in tandem with mass spectrometry since they report concentrations of precursors and quantify our desired product. However, gas chromatography would require the derivatization of progesterone so it could be vaporized during the mobile phase. We determined that we should use liquid chromatography with mass spectrometry due to the convenience of the solid phase extraction system. Solid phase extraction utilizes the affinity of mobile phase solutes for a solid in its stationary phase to separate a mixture into its components by means of a cartridge. We would also use affinity column chromatography with progesterone-antibody beads, or ELISA for a more specific detection of progesterone. These methods would require progesterone antibodies and more preparation, but allow for immediate on site results.
DOSAGE
Our project requires a method for a consumer to obtain a reliable dose of contraceptive. Initially, we considered a galactose inducible progesterone biosynthesis pathway so we could establish a concentration of substrate to product curve. We abandoned this idea after learning that galactose inducibility is repressed by glucose which is present in the fuel source. We then considered methods of ceasing production of progesterone by use of a sensor that would inhibit the pathway at a specific concentration of progesterone.
Progesterone Receptor
The human progesterone receptor has 3 common isoforms, one of which is inherently cytosolic and translocates into the nucleus to affect gene expression after binding to progesterone. We considered using this action to initiate an apoptosis event upon binding of progesterone in the cell. This idea had some problems; the natural binding affinity for these receptors was much too high for our purposes and would result in apoptosis when the cell had accumulated ~0.05% of our target progesterone concentration of 20mg. This concentration is the required dosage for progesterone to suppress ovulation when taken sublingually. Changing the binding affinity of a cell receptor would require a large effort to intelligently design a new receptor or, more likely, a large library of random mutant receptors to screen. This idea was discounted fully after we discovered that the innate activity of the receptor tended to disrupt the apoptosis pathway.
Riboswitch
We still liked the idea of creating a sensor that could inform the consumer when the desired concentration of progesterone was present in the culture. From our mentor, McKenna Hicks, we learned about riboswitches, due to her extensive research and creation of the nucleotide-based sensors. We were informed that the guided mutagenesis process for a protein-based receptor was comparatively more labor-intensive anyways than for the sensors that could be created with riboswitches.
Riboswitches are composed of short sequences of nucleotides made up of an aptamer, which is a ligand-binding domain made up of nucleic acids, and the expression platform that can control gene-regulatory functions. These aptamers can be tested in vitro for progesterone specificity, whereas proteins require synthesis in vivo. A paper by Contreras Jimnenez et al. (2015) described several in vitro-derived aptamers specific for progesterone [28]. Unfortunately, like the progesterone receptor, these aptamers are also three orders of magnitude too sensitive for our desired concentration, but have much shorter sequences - an average of 51 bp compared to 933 bp for human progesterone receptor isoform B (hPR-B) [28,29]. Mutating these short sequences and isolating variants would be much easier than mutating a larger protein receptor. We found that the systematic evolution of ligands by exponential enrichment (SELEX) method follows a similar logic to directed evolution, and is intended for selecting RNA aptamers [30].
For these reasons, we looked into the viability of creating a progesterone-specific RNA aptamer that could be used in conjunction with a riboswitch expression platform to affect the expression of a reporter gene. Our research uncovered a previous iGEM BioBrick (BBa_K1913011) that used a trans-functioning riboswitch to indirectly affect the expression of an RFP reporter gene directly affecting the expression of a TutR inverter protein as well as another paper describing the in-vitro characterization of several progesterone-specific RNA aptamers. We attempted to mimic the same TutR inverter system as in the previously described BioBrick until we found another paper describing a more fitting option.
PoPPY-switch
The expression platform of the hammerhead ribozyme has been altered to be more accommodating to the attachment of arbitrary RNA aptamers, giving us the modularity and cross-domain function needed to create our sensor. With this new information, our project then became the creation of a novel single gene reporter system.
Win and Smolke (2007) published research on the hammerhead ribozyme, which works across varying domains, where they increased the modularity of the expression platform by adding two small nucleotide sequences labeled as the switching and competing strands as seen in Figure 6[31]. This system allows for the combination of arbitrary aptamers with the hammerhead expression platform. In its active state, the hammerhead ribozyme causes a cleavage event in the 3’ untranslated region (UTR) of the transcribed mRNA, resulting in accelerated degradation of the mRNA strand. Depending on the location and sequence of the switching and competing strands, the hammerhead ribozyme can be either constitutively active or inactive, with ligand binding switching it to the opposite state.
The riboswitch will consist of the hammerhead ribozyme expression platform, a progesterone-specific aptamer, and switching/competing strands oriented so that the ribozyme is constitutively active and then inactivated by rising progesterone concentrations.

Figure 5: Graphic adapted from Win and Smolke (2007) showing the modularity of the hammerhead ribozyme (HHR) with arbitrary aptamers. Different aptamers (dashed boxes) can be used with the same expression platform while still showing a change of activity upon binding of a specific ligand. We will use this system with our progesterone aptamer.
For our purposes, hrGFP will be the transcribed reporter gene. Progesterone bound to the aptamer will halt the degradation of hrGFP mRNA and allow our Y. lipolytica cells to produce GFP product. In this way we can detect the presence of progesterone by the fluorescence of GFP when progesterone is bound, and we can use SELEX to create aptamers that are less sensitive to progesterone and will show visible fluorescence when the culture reaches medically useful levels.
We need to engineer the riboswitch into the genome so consumers can always have a visual representation of when their culture is ready for use. Cory Schwartz from UC Irvine donated the pHR-D17-hrGFP plasmid to us, which has homology arms to the Y. lipolytica genome. The plasmid can integrate into the D17 pseudogene of the Y. lipolytica genome by homologous recombination and has a constitutive expression of GFP [22]. We will add our short riboswitch sequence (219-222 bp, depending on the specific aptamer) in the 3’ UTR of the hrGFP reporter of the plasmid; this creates a one-gene reporter system contained within a plasmid with ampR and ura3 selectable markers and an hrGFP reporter linked to a progesterone-specific riboswitch.

Figure 6: Design of our one-gene riboswitch reporting system. The absence of progesterone allows for continued activation of the hammerhead ribozyme, which cleaves the hrGFP mRNA at the 3’ end and causes accelerated degradation of the mRNA strand. In the presence of progesterone, the hammerhead ribozyme is inactivated and translation occurs normally. The frequency of translation events will be tied to the frequency of ligand binding events, allowing for concentration-dependent levels of fluorescence.
FUTURE WORK
Progesterone's direct precursor, pregnenolone, also has many beneficial aspects. It is the precursor to all steroid hormones in the body, such as cortisol and aldosterone. Cortisol and aldosterone are adrenal cortex steroid hormones. Cortisol regulates carbohydrate metabolism and has an anti-inflammatory effect on the body. Aldosterone plays a huge role in cardiovascular health, and helps maintain blood pressure and regulates the salt and water balance in the body. The pathway design we have created allows for the future creation of many beneficial steroid drugs by simply adding a few more genes.
Future optimizations for PoPPY-switch could include changing the location of our riboswitch so that high progesterone concentrations directly inhibit the progesterone production pathway creating an innate dosage-control mechanism. Another avenue of improvement would be to use the same riboswitch system, but with a constitutively inactive cleavage that shuts down the production of a survival-necessary gene upon binding to progesterone. The resulting system would then use senescence as an off-switch for progesterone production.
- U.S. Food & Drug Administration (2011). GRAS Notices - GRN No. 355.
- Groenewald, M., Boekhout, T., Neuvéglise, C., Gaillardin, C., Dijick, P. W. M. v., and Wyss, M. (2014). Yarrowia lipolytica: Safety assessment of an oleaginous yeast with a great industrial potential. Critical Reviews in Microbiology 40, 187-206.
- Madzak, C., Gaillardin, C., and Beckerich, J.-M. (2004). Heterologous protein expression and secretion in the non-conventional yeast Yarrowia lipolytica: a review. Journal of Biotechnology 109, 63-81.
- Roostita, R. and Fleet, G.H. (1996). Growth of yeasts in milk and associated changes to milk composition. International Journal of Food Microbiology 31, 205-219.
- Fabiszewska, A. U., Kotryba, D., and Nowak, D. (2015). Assortment of carbon sources in medium for Yarrowia lipolytica lipase production: A statistical approach Annals of Microbiology 65, 1495-1503.
- Jang, I.-S., Yu, B. J., Jegal, J., Lee, J. Y. (2018). Improving the efficiency of homologous recombination by chemical and biological approaches in Yarrowia lipolytica PLOS
- What is Progesterone?
- Apgar, B. S., and Greenberg, G. M. (2000). Using Progestins in Clinical Practice. AFP 62, 1839–1846.
- Campagnoli, C., Clavel-Chapelon, F., Kaaks, R., Peris, C., and Berrino, F. (2005). Progestins and progesterone in hormone replacement therapy and the risk of breast cancerThe Journal of Steroid Biochemistry and Molecular Biology 96, 95-108.
- Kanehisa,M. and Goto, S. (2000). KEGG: new perspectives on genomes, pathways, diseases and drugs Nucleic Acids Research 28, 27-30.
- Money, N.P, Watkinson, S.C., Boddy, L. (2016). Chapter 2 - Fungal Cell Biology and Development. In The Fungi (Boston: Academic Press). 3rd edn., 37–66.
- Duport, C., Spagnoli, R., Degryse, E., and Pompon, D. (1997). Self-sufficient biosynthesis of pregnenelone and progesterone in engineered yeast. Nature Biotechnology 16, 186–189
- Waterham, H.R., Wanders, R. J. A. (2000). Biochemical and genetic aspects of 7-dehydrocholesterol reductase and Smith-Lemli-Opitz syndrome Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1529, 340-356.
- Grinberg, A.V., Hannenemann, F., Schiffler, B., Müller, J., Heinemann, U., Bernhardt, R. (2000). Adrenodoxin: Structure, stability, and electron transfer properties Proteins: Structure, Function, and Bioinformatics 40, 590–612.
- Koritz, S. B. (1964). The conversion of pregnenolone to progesterone by small particles from rat adrenal* Biochemistry 3, 1098–1102.
- Hoover, D. M. and Lubkowski, J. (2002). DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Research 30, e43
- Curran, K. A., Morse, N. J., Markham, K. A., Wagman, A. M., Gupta, A., and Alper, H. S. (2015). Short synthetic terminators for improved heterologous gene expression in yeast. ACS Synthetic Biology 4, 824–832.
- Celinska, E., Ledesma-Amaro, R., Larroude, M., Rossignol, T., Pauthenier, C., and Nicaud, J. (2017). Golden Gate Assembly system dedicated to complex pathway manipulation in Yarrowia lipolytica. Microbial Biotechnology 10, 450–455.
- New England BioLabs (n.d.). Golden gate assembly.
- Summers, D. K. (1991). The kinetics of plasmid loss. Trends in Biotechnology 9, 273–278.
- Wong, L., Engel, J., Jin, E., Holdridge, B., and Xu, P. (2017). YaliBricks, a versatile genetic toolkit for streamlined and rapid pathway engineering in yarrowia lipolytica. Metabolic Engineering Communications 5, 68–77.
- Schwartz, C., Shabbir-Hussain, M., Frogue, K., Blenner, M., and Wheeldon, I. (2017). Standardized markerless gene integration for pathway engineering in Yarrowia lipolytica. ACS Synthetic Biology 6, 402–409.
- Noble, C., Adlam, B., Church, G. M., Esvelt, K. M., and Nowak, M. A. (2018). Current CRISPR gene drive systems are likely to be highly invasive in wild populations. eLife
- Riveline, D. and Nurse, P. (2009). Injecting yeast. Nature Methods 6, 513–514.
- Verbeke, J., Beopoulos, A., and Nicaud, J.-M. (2013). Efficient homologous recom- bination with short length flanking fragments in ku70 deficient Yarrowia lipolytica strains. Biotechnology Letters 35, 571–576.
- Watson, A. T., Garcia, V., Bone, N., Carr, A. M., and Armstrong, J. (2008). Gene tagging and gene replacement using recombinase-mediated cassette exchange in schizosaccharomyces pombe. Gene 407, 63–74.
- Juchheim, A. M. (2015). Plasmids 101: Cre-lox.
- Contreras Jimenez, G., Eissa, S., Ng, A., Alhadrami, H., Zourob, M., and Siaj, M. (2015). Aptamer-Based Label-Free Impedimetric Biosensor for Detection of Progesterone. Analytical Chemistry 87, 1075–1082.
- UniProt (n.d.). UniProtKB - P06401 (PRGR human).
- Ellington, A. D. and Szostak, J. W. (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822.
- Win, M. N. and Smolke, C. D. (2007). A modular and extensible RNA-based gene- regulatory platform for engineering cellular function. Proceedings of the National Academy of Sciences 104, 14283–14288.