Team:SKLMT-China/Model Process
Background
We have built a promoter library of inner promoters with various strength in
1.To facilitate the promoter transformation of pseudomonas fluorescence in new site;
2.To reveal the correlation between promoter weights and their strength;
4.To improve the parameter setting of the promoter strength prediction in the species outside the large intestine with the method of position weight;
5.To provide reference for other microbe promoter strength prediction modeling.
Assumptions
1. It is assumed that the promoter strength is only related to the core DNA sequence.In other words, the DNA sequence can uniquely determine the promoter strength.
2. It is assumed that the laboratory environment has no impact on the test results.In other words, our test data can fully reflect the promoter strength.
3.It is assumed that there is a simple monotonic relationship between the obtained fluorescence data and the promoter strength: the larger the fluorescence value is, the stronger the promoter can be considered.
Modelling Process
DNA Sequence Encoding
The 5’-3’ region includes 70bp DNA information in each promoter. Currently, we have got 25 promoter sequences. We need to encode these 70 bases in order to establish a mathematical model. Notice that there’re only four types of bases(A,T,C and G),and calculating the numbers of each type is not enough to reflect interaction between adjacent bases.[2] Thus, we consider 3 continuous bases
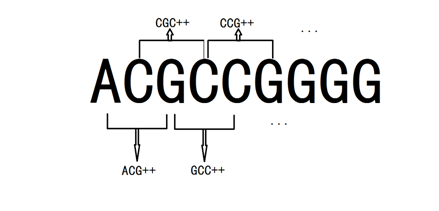
We encode each three-base sequence with quaternary number(A,T,C,G represents quaternary number 0,1,2,3 ), and by running a C++ program (the whole code is shown in appendix), we get a vector with 64 dimensions from AAA to GGG, which calculates the frequency of appearance for each type in DNA sequence.
After encoding, all DNA sequences are transformed into a vector with 64 dimensions. The entire data is stored in the document
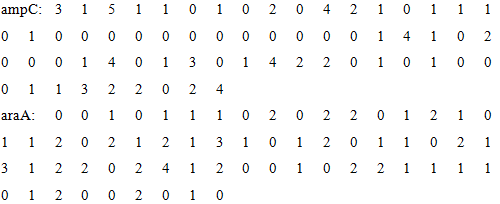
At the end of the encoding step, we get an 25*64 matrix which reflects the features of these DNA sequences of each promoters. This encoding method has deep internal connection with the characteristics of DNA sequence, and it simultaneously contains the sequential relationship and details of DNA sequence. With this matrix, a mathematical model can be established.
Model establishment and solution
In order to simplify the problem, we assume that the connect between matrix and promoter strength is linear, in other words, the vector \({x}\) exists when equation \({y=Ax}\) is satisfied.
Now, through a lot of experiments, we have measured a total of 24 values, and all twenty-two valid values are shown below. Each data is the average of the results of three parallel experiments.
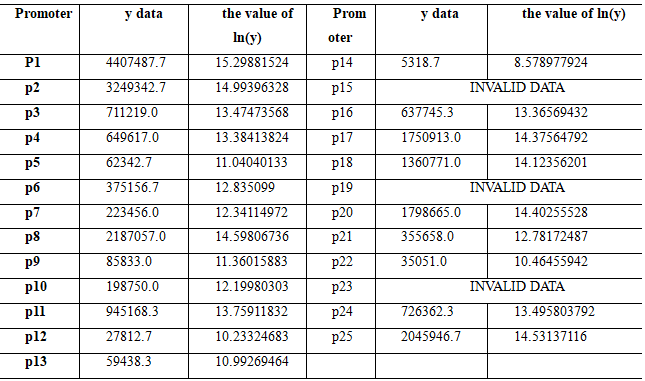
The gap in original data is too wide, so we take the natural log of the original data as the value of the vector . Now, what we're going to do is to invert the linear relationship vector \({x}\) by taking the linear transformation matrix \({A}\) and the resulting vector \({y}\).\({A,x}\) and \({y}\)satisfy the following equation: \({y=Ax}\),or \({x=A-1y}\).
This is a linear mapping of 64 variables but there are only 22 data points. In other words, this is an underdetermined equation which cannot be solved accurately by solving inhomogeneous linear equations. The sparse solution algorithm of underdetermined equations based on iteration3 will be adopted in the following part.
This is an effective algorithm to restore deterministic signals with fewer premeasured values.
Our goal is to improve the precision of signal reduction as much as possible. Consider the following optimization problem:
$$min \left \| x \right \|_{0}$$ $$s.t.\; \; \; y=Ax$$