Team:UFlorida/Demonstrate

Demonstrate
The first step of our system design was to delete naturally occurring genes in E. coli Nissle 1917 that encode for interfering metabolic enzymes (ldhA, adhE, frdA, pta ). This would allow subsequent insertion of the butyrate gene and actually produce significant yield in a strain unable to do so before. Other iGEM teams have attempted to engineer butyrate production but were unsuccessful due to lacking key enzymes or accurate gene sequences. We were not able to assemble an RFC10-compliant butyrate pathway BioBrick. However, we have properly introduced butyrate production in two E. coli strains, proving our engineered system works. We also demonstrated that deleting the ldhA gene in E. coli Nissle did influence the carbon flux in a manner that increased yields of other by-products, such as ethanol, in lieu of lactate production.
It follows that in this deletion strain and in subsequent deletion strains, a completely NADH-dependent plasmid-based butyrate production pathway could be functional and subjected to further metabolic engineering by directed evolution. If an iGEM team were to create this pathway, either de novo, cloning, or by adding the ter gene to BBa_K1587004, they would be able to use our deletion strain as a chassis for their part. Finally, metabolic engineering of E. coli Nissle 1917 to produce other by-products could potentially make use of our deletion strain, which will be available to other iGEM teams by request.
As a last ditch attempt to produce a butyrate-producing part, we attempted to add the Ter gene, which was given to us as a gift from Dr. Shanmugam, to our system. This gene contains the same amino acid sequence (but different codon optimization) than our submitted Ter part (BBa_K2677001). Placed under control of the pBAD promoter (BBa_I0500) downstream of BBa_K1587004’s terminator, which contains 4 out of the 5 enzymes needed to produce butyrate in E. coli, would contain a complete butyrate pathway. Unfortunately, the AraC-pBAD-Ter fragment cloned in the wrong orientation into the XbaI and SpeI sites, and therefore it is not RFC10 compliant. Given more time and resources, we might have found a colony with the correct orientation; however, in an attempt to prove that our engineered system works, we will continue to show data for this plasmid, which will be made publicly available to any team that requests it. We will refer to this plasmid as the “butyrate pathway part” from here on.
BEM3
BEM3 was a strain of E. coli used to test the function of our part, which is intended for use in our deletion strain of E. coli Nissle 1917. BEM3 has several pertinent genes deleted, including ldhA and adhE. It can grow anaerobically only if it has been transformed with a plasmid that contains a pathway for it to produce specific metabolites such as butyrate. We transformed BEM3 with this butyrate path part. We observed the following optical densities at the same time point after 24 hours of anaerobic incubation at 37 degrees:
Anaerobic Incubation at 37 C
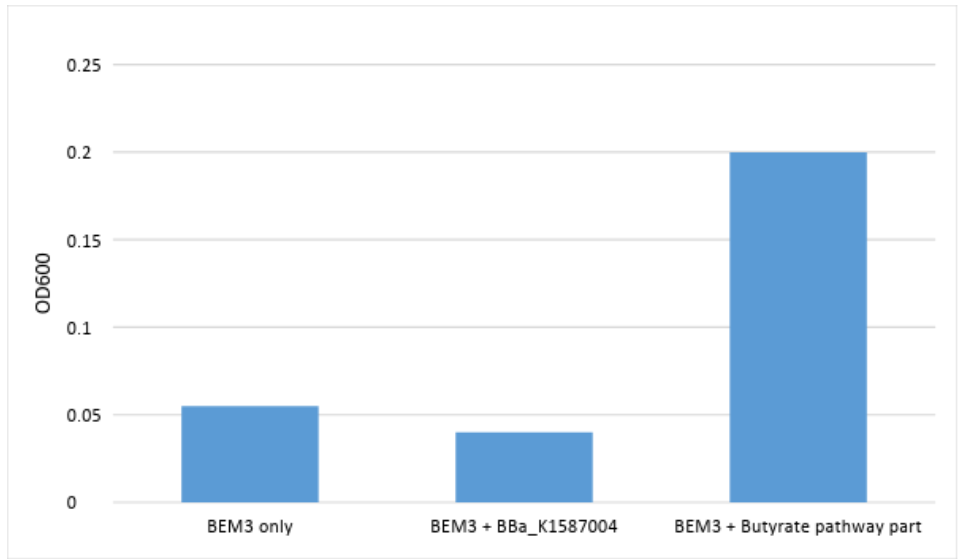
From the measured OD’s, we can conclude that our part is functionally providing a metabolic pathway for BEM3 to survive anaerobically. BBa_K1587004 alone does not allow the BEM3 strain to grow anaerobically because it does not contain Ter, an NADH-dependent enzyme, and is therefore not redox balanced for butyrate production. Ccr is not sufficient for BBa_K1587004, as it is an NADPH-dependent enzyme. Our part increased the optical density to four times what was observed in the other groups, suggesting it is providing a pathway for BEM3 to produce butyrate and survive anaerobically, something that BBa_K1587004 alone did not do.