Team:SUSTech Shenzhen/Engineered cells
(1) Secreting cell line --- L-Wnt3A cell line
Characteristics:
This cell line can secret the signal protein -- Wnt3a protein, and it is derived from the L-M cell, which is transfected with a Wnt-3A expression vector and stable clones, further selected in medium containing G418. (Sources from ATCC: antibiotic selection, ref).
Overview:
We firstly use the second-generation Lentiviral vectors containing three separate plasmids to produce lentivirus. Lwnt3A are transfected by lentivirus that contains Cas9-mCherry gene and gRNA sequences successively. After that, we confirmed the transfection efficiency of target genes via flow cytometry and verified the same Wnt secretion capability of genetically modified cells by Western Blot. To optimize our Wnt-secreting cell lines, we selected single clonal cells and confirmed their knockout efficiency through FACS after coculture with the reporter cell.
Plasmids construction:
In our project, we introduce the Cas9 system to the L-Wnt3A cell line, and it can be easily manipulated in gene level. We design primers to amplify this whole sequence and use Gibson Assembly to ligate it into lentivirus backbone, so it can be packaged into the Lentiviral genome and then stably integrated into the L-Wnt3A cell line.

Figure 1. Cas9 construct in Lentiviral backbone
In this plasmid (Fig1), because Cas9 gene is fused by a mCherry fluorescence gene through a T2A, they will be expressed together. Therefore, we can select out the cells that successfully receive Cas9 and express it.

Figure2. WNT secretion pathway
In order to perform verification of our engineered system, we introduced alterations in secretion pathway using CRISPR-Cas9 knockout(Protocols). Firstly, we chose WNT3A gene and two wnt3a maturation-related gene, PORCN and WLS. As Fig2 shows, porcupine O-acyltransferase (PORCN) locates in ER, functioning as an essential enzyme to modify the Wnt3A protein. And Wntless WNT ligand secretion mediator (WLS) is another essential protein that mediate transportation of Wnt3a from Golgi to plasma membrane. We search the sequence of these three genes in NCBI and design the single guide RNAs for them according to the principle of CRISPR-Cas9, also we included a random sequence that did not target any sequence

Figure3. Constructs of gRNA of known Wnt secretion genes and non-target
And we inserted these gRNAs into the Lentiviral backbone, and the transcription of gRNA is under control of U6 promoter. Besides, in order to select out the successfully transfected cells, we also added a puromycin resistance gene following EF-1α promoter. Totally, we have four Lentiviral-sgRNA plasmids. (Fig3) All the guide RNA sequence together with their scaffold have been added to our parts.
After insertion of gRNA, we did Sanger sequencing, and results shown we were success. Example of gRNA of WNT is shown in Fig4.

Figure4. Alignment of designed plasmid and product plasmid of Lentiviral-sgWNT.
Functional verifications:
After construction of all these plasmids, we perform cell experiments to verify our results and continue to get stable secretion cell line for our Double Emulsion System.
1. Cell transfection by Lentivirus carrying Cas9 gene.
In order to obtain a donor cell line which can be easily manipulated in gene level, we consider introducing the CRISPR-Cas9 system to original cell line. After virus production, we infect the cell with Cas9-mCherry carrying lentivirus in safe condition. Due the expression of Cas9 and mCherry are coupled, we use FACS to sort out the successfully transfected cells. Results are shown in Fig5.

Figure5. L-Wnt3A cell before and after transfection with the Cas9-mCherry by lentivirus. mCherry fluorescence shift means the Cas9-mCherry has been stably expressed in this new cell line.
2. Western Blot
To ensure the cell line can normally secret Wnt3A protein and to quantity of it, we used Western Blot(WB) to detect the concentration of the protein in the supernatant of adherent cells and suspension cell. The results in Fig6 show 72h incubation can produce about 200 to 400ng/mL wnt3a, which is good. The other bands in 50KD can be due to the non-specificity of antibodies.
The 12h results show no bands, because the medium is from 10cm dish with about 107 cells, so each cell possesses much larger microenvironment. On the other hand, the microenvironment in a droplet is much smaller than that in the culture dish. So this result makes sense and illustrate that secretion cells can secret Wnt3a within droplets in a shorter time indirectly.

Figure 6. Detection of the Wnt in the cytoplasm and suspend in the L-Wnt3A cell culture by Western Blot. (Lane1, 11: Ladder; Lane 2, 3, 4, 5: standard concentration gradient of Wnt protein; Lane 7, 8, 9: the supernatants of the cell with different culturing time; lane 10: cells cultured by the suspension method, 24h)
3. Cell transfection by the Lentivirus carrying gRNA (WLS, PORCN, WNT, and NON-target) These cell lines is further transfected with the different gRNA, and selected out by the Puromycin. And the resultscan be shown with coculture with receptor cells.
Optimization strategy:
Single clone selection
In order to minimalize the variables, we wanted to obtain a homogenous cell line, so, we decided to do the single clonal selection for the L-Wnt3A cas9 cell line and L-Wnt3A Cas9-KO cell line. We use limited dilution to decrease the density of the cells to ensure one well of 96-well plate had almost one cell., and culture the cell in 96-well plate, then transfer to larger one. (Fig7) In this way, we got a new cell line which was derived from one single cell and possessed the same genotype as well as phenotype.

Figure7. Single clone strategy.
(2) Responding cell line --- HEK293R cell line:
Characteristics:
HEK293R is a sub cell line of HEK293. It can easily to be transfected with plasmids or Lentivirus. Besides, the Wnt protein from mouse is a homolog of that from human, and the human receptor of Wnt protein can be targeted by the Wnt protein secreted from mouse. So, the origin difference of two cell line does not affect too much.
Overview:
In order to detect the Wnt signal from Wnt secreting cells after gene knockout, we should choose a method to quantify the resulting effects. One is to use immunofluorescence to detect the concentration of Wnt3a while the other is to use a reporter cell line to detect the Wnt3a signal. But for immunofluorescence, it is unlikely to detect concentration of Wnt3a in each microdroplet. While utilizing a reporter cell as the detector in a single droplet is superior. So, we begin to construct Wnt signaling reporter cell lines.
Plasmids construction:
In our project, we design two kinds of cell line to receive the signal from the L-Wnt3A cell line and further give response according to the basic principle of Wnt signaling pathway. One of them is the TCF-GFP cell line. TCF is a specific promotor functioning at the end of canonical Wnt signaling pathway, it can be regulated by the signal from cytosol, such as beta-catenin, and then activate the transcription of the downstream genes, there we make the GFP reporter as one of the downstream genes. (Fig8)

Figure8. Wnt signaling pathway modified by a TCF-GFP construct.
And we construct the lentiviral vector, which contains three TCF transcription response elements (TREs), a TATA box, and a GFP gene. We include three TCF TREs to enhance the response. In the construct Fig9, TCF actually represents three TCF TREs and a TATA box.

Figure9. Construct of TCF-GFP in lentiviral backbone
On the other hand, the downstream effect of Wnt signaling pathway can also be the accumulation of β-catenin in cytosol.() Therefore, if we can add a reporter to reflect the quantity of β-catenin, we can know the response strength of Wnt signaling initiated by Wnt ligand. So, the strategy is fusing a GFP with β-catenin. (Fig10) Then, we can estimate the quantity of β-catenin according to green fluorescence intensity.

Figure10. Wnt signaling pathway modified by a GFP-β-catenin construct.
At the beginning, we amplify the β-catenin sequence and insert it into the lentivirus backbone under the control of EF-1α promoter.(Fig11) Besides, we introduce an EGFP to fuse with β-catenin. Sanger sequencing results show we have successfully constructed the plasmid. (Fig12) and we have integrated TCF-GFP construct into pSB1C3 as one of our improved parts.

Figure11. Construct of the GFP-β-catenin in lentiviral backbone.

Figure12. alignment of designed plasmid and sequencing results of product plasmid.
Functional verifications:
After construction of these two reporter plasmids, we perform cell experiments to verify that whether they can work or not, and continuously, we construct the stable reception cell lines for our Double Emulsion System.
1. Cell transfection by lentivirus that carry GFP-β-catenin or TCF-GFP.
After packaging the lentivirus, we infect the original HEK293R cell line under the safety rules.
For the cells transfected by GFP-β-catenin virus, under the fluorescence microscope, we can observe cells show green fluorescence at a basal level without adding any extracellular signal like Wnt. That is because β-catenin is always expressed but they go to degradation mediated by disruption complex. () Therefore, they should maintain a basal protein level, which leads to a basal green fluorescence in our GFP-β-catenin cell line. While when Wnt signal comes, the green fluorescence intensity should increase to a higher level. For optimization, we intend to enlarge the gap by sorting out the cells with the lowest green fluorescence. After sorting, under fluorescence microscope, we can see the green fluorescence is relatively lower, but it still not so ideal. (Fig13)

Figure13. Fluorescence images of GFP-β-catenin cell line after low fluorescence sorting.
For the cells transfected by TCF-GFP virus, we have successfully got a stable transfected cell line. At this time, we introduce a drug named Chir99021, one kind of inhibitors of disruption complex. In other words, Chir99021 activates Wnt signaling pathway with lower cost compared to pure Wnt3a protein ligand. After adding 3μM Chir99021 or 200ng/mL of Wnt3a, TCF-GFP cells give out green fluorescence from dark after specific incubation time. (Fig14)

Figure14. Fluorescence images of TCF-GFP cell lines under specific concentration of Chir99021 or pure Wnt3a protein.
Comparing these two reporter systems of our receptor cells, GFP-β-catenin and TCF-GFP, we can draw some conclusions. The response time of GFP-β-catenin cell line is shorter, but it has a basal fluorescence which makes it’s hard to quantify the differential fluorescence between normal condition and stimulated condition. While for the TCF-GFP cell line, the activation is from 0(no fluorescence) to 1(fluorescence), however, from the Wnt ligand to the target gene expression, longer time is needed. (Table1)
Table1. Comparison of two reporter system strategies.

2. Selection of the cells with strong response ability by coculture and drug treatment.
In order to satisfy the requirement, we need to select out a high effective reporter cell line, in other words, the receptor cell should have a distinct change in fluorescence strength in a short time after receiving extracellular signal. Then we use TCF-GFP cell line.
Thus, our initial strategy is to select the cells by flow cytometry, through the maximum change of the fluorescence strength between normal condition and drug treatment.
And we consider three different ways (Chir99021 stimulation; pure Wnt3A protein activation; coculture with the L-Wnt3A secretion cell) to select the ideal receptor cell. Chir99021 stimulation is shown in Fig15, cells located in very right region obviously have larger changes and they are sorted out.

Figure15. TCF-GFP cells were treated by 5 μM Chir99021 in 24h. The first diagram is control group, and second diagram is experimental group.
Optimization strategy:
In fact, when we constructed the reporter cell lines, we faced lots of problems.
The first is the heterogeneity of the cell line, from the flow cytometer analysis of the reporter cell line, we can see there are various cell types with different GFP intensity, (Fig16) which discourage us. After talking with professors, we know that when we use virus infection to get stable cell line, there is possibility for one cell to be integrated random copies of construct. And then we use the single clone selection, which described above, to solve this problem.
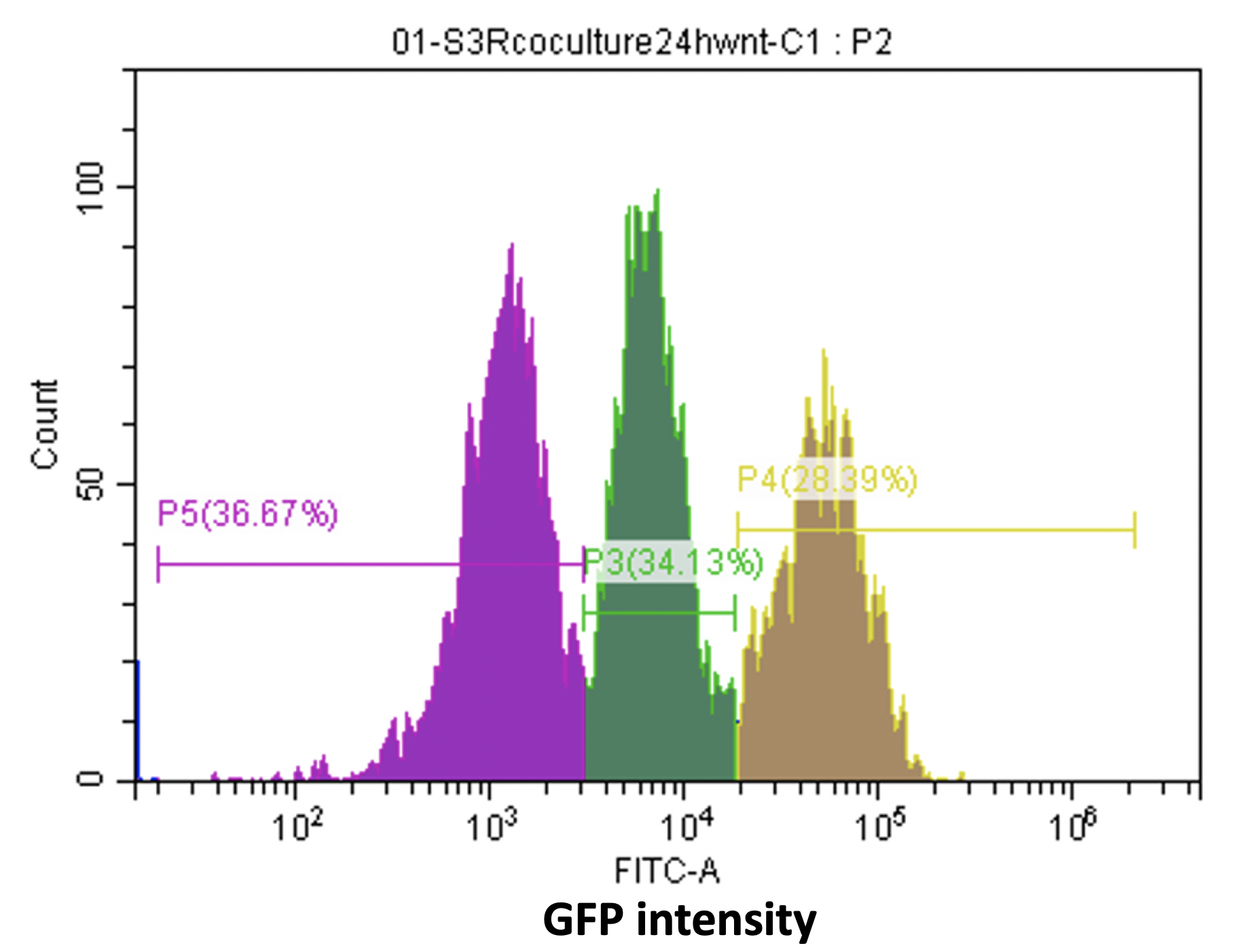
Figure16. Various cell types with different GFP intensity in the reporter cell line.
The following problem is how to increase the efficiency of the receptor cell line further. We do need a sharp change in fluorescence strength of reporter cell line in several hours. As mentioned above, cells can obtain different copy numbers of reporter construct, so the single clones we grow are underwent copy number investigation to select out the clones with the highest copy number, which must have the highest response ability.
Also, we are considering optimizing the construction of reporter cell line from the perspective of signaling pathway. That means we adjust the components expression level in signaling pathway to optimize the path from Wnt ligand to Wnt target. For example, we overexpress the membrane receptor of Wnt signal to increase the sensitivity to Wnt signal hopefully. Based on this hypothesis, we use our modeling, kinetic model of Wnt signaling pathway, to find out how to optimize the pathway. The results surprise us, explore it in here.
(3) Coculture of two cell lines - Donor & Receptor:
After the donor cell line and receptor cell line are fully constructed, we perform coculture to test the interaction and communication between them. So, we have L-Wnt3A-Cas9-mCherry-KO cell lines, which contains three genes knock out, PORCN, WLS and WNT, and also a NON-target gRNA. For the receptor cell, we have 293R-TCF-GFP cell line. The results of coculture are shown in Fig17, we can clearly see the red fluorescence alternating with green fluorescence in NON-target group and only red fluorescence in secretion gene-KO groups.

Figure17. Coculture of 293R TCF-GFP cells with L-Wnt3A-Cas9-mCherry-KO cells.
©2018 SUSTech_Shenzhen
